Day 8: Workflow Wrappers & Web Platforms - Streamlining Metagenome Analysis
Day 8: Workflow Wrappers & Web Platforms
| Estimated Time: 4-6 hours | Difficulty: Beginner to Intermediate | Prerequisites: Understanding of Days 1-7 concepts |
📚 Table of Contents
- Introduction
- Part I: Workflow Wrappers
- Part II: Web-Based Platforms
- Part III: Choosing the Right Tool
- Best Practices
🎯 Introduction
You’ve learned the individual tools. Now let’s automate everything.
The Challenge
Days 1-7 taught you:
- Quality control
- Assembly
- Binning
- Annotation
- Comparative analysis
But running each tool separately is:
- ❌ Time-consuming
- ❌ Error-prone
- ❌ Hard to reproduce
- ❌ Requires deep Linux knowledge
The Solution
Workflow wrappers = Pre-built pipelines that automate everything
Web platforms = Run analyses in your browser (no installation!)
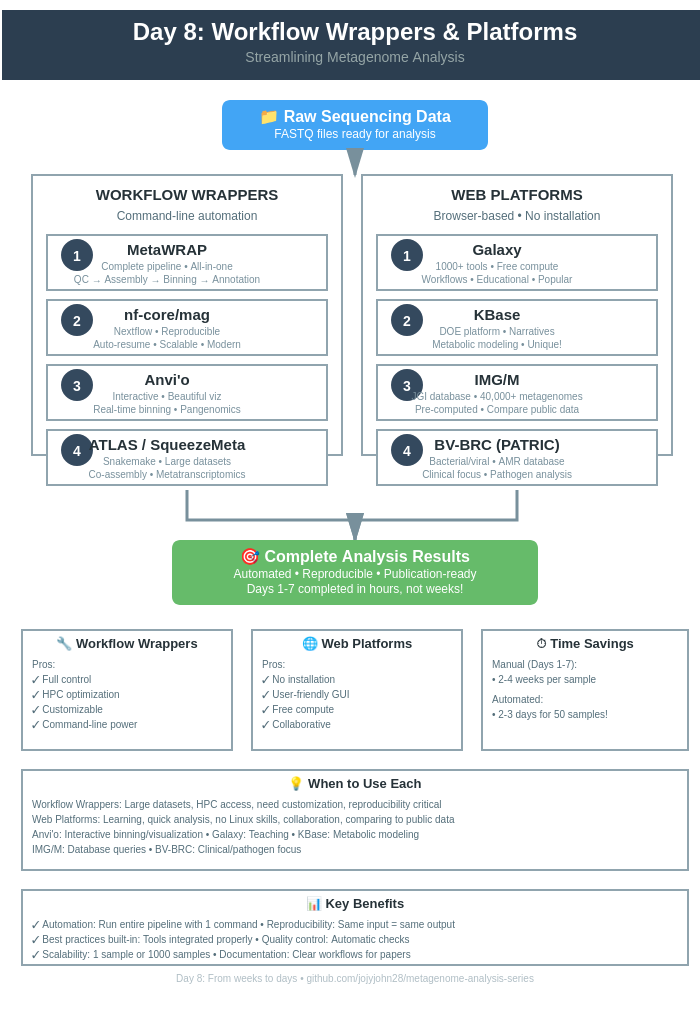
🔧 Part I: Workflow Wrappers
Overview
| Tool | Type | Best For |
|---|---|---|
| MetaWRAP | All-in-one | Complete workflows, HPC |
| nf-core/mag | Nextflow | Reproducible, scalable |
| Anvi’o | Interactive | Visualization + analysis |
| ATLAS | Snakemake | Large datasets, metatranscriptomics |
| SqueezeMeta | Integrated | Co-assembly, functional annotation |
🎁 1. MetaWRAP - Complete Metagenome Pipeline
MetaWRAP wraps Days 1-7 into simple commands.
Installation
# Create conda environment
conda create -n metawrap-env python=2.7
conda activate metawrap-env
# Install MetaWRAP
conda install -c ursky metawrap-mg
# Download databases (WARNING: ~300 GB!)
metaWRAP_db_setup.sh
Complete Workflow
please use metaWrap_complete_workflow.sh from Day 8 →
MetaWRAP Modules
Available modules:
# Quality control
metawrap read_qc
# Assembly
metawrap assembly --metaspades # or --megahit
# Binning
metawrap binning --metabat2 --maxbin2 --concoct
# Refinement
metawrap bin_refinement -c 50 -x 10
# Reassembly
metawrap reassemble_bins
# Quantification
metawrap quant_bins
# Classification
metawrap classify_bins
# Annotation
metawrap annotate_bins
# Bin visualization
metawrap blobology
Advantages
✅ All-in-one solution
✅ Integrates multiple tools
✅ Automatic bin refinement
✅ Good documentation
Disadvantages
❌ Large database requirements (~300 GB)
❌ Python 2.7 dependency
❌ Can be slow
❌ Less flexible than individual tools
🔁 2. nf-core/mag - Nextflow Pipeline
Nextflow = Modern workflow manager (reproducible, scalable)
Installation
# Install Nextflow
curl -s https://get.nextflow.io | bash
mv nextflow ~/bin/
# Or via conda
conda install -c bioconda nextflow
Running nf-core/mag
# Basic run
nextflow run nf-core/mag \
--input samplesheet.csv \
--outdir results \
-profile docker
# Full options
nextflow run nf-core/mag \
--input samplesheet.csv \
--outdir results \
--skip_spades \
--megahit_fix_cpu_1 \
--min_contig_size 1500 \
--min_length_unbinned_contigs 1000000 \
--max_unbinned_contigs 100 \
--busco_db bacteria_odb10 \
-profile docker,test
Sample Sheet Format
sample,group,short_reads_1,short_reads_2,long_reads
sample1,group1,s1_R1.fastq.gz,s1_R2.fastq.gz,
sample2,group1,s2_R1.fastq.gz,s2_R2.fastq.gz,s2_long.fastq.gz
sample3,group2,s3_R1.fastq.gz,s3_R2.fastq.gz,
nf-core/mag Features
✅ Modern workflow engine
✅ Automatic resume (restart from failures)
✅ Multiple assemblers (MEGAHIT, SPAdes)
✅ Multiple binners (MetaBAT2, MaxBin2, CONCOCT)
✅ BUSCO quality checks
✅ GTDB-Tk classification
✅ MultiQC reports
Output Structure
results/
├── QC/
│ ├── fastp/
│ └── fastqc/
├── Assembly/
│ ├── MEGAHIT/
│ └── SPAdes/
├── Binning/
│ ├── MetaBAT2/
│ ├── MaxBin2/
│ └── bins_summary.tsv
├── GenomeBinning/
│ ├── bins/
│ └── checkm2_results/
├── Taxonomy/
│ └── GTDB-Tk/
└── MultiQC/
└── multiqc_report.html
🖥️ 3. Anvi’o - Interactive Metagenomics
Anvi’o = Analysis + Visualization platform
Installation
conda create -y -n anvio-8 python=3.10
conda activate anvio-8
conda install -y -c conda-forge -c bioconda anvio=8
anvi-self-test
Quick Workflow
# 1. Create contigs database
anvi-gen-contigs-database -f contigs.fa -o contigs.db
# 2. Annotate
anvi-run-hmms -c contigs.db -T 8
anvi-run-ncbi-cogs -c contigs.db -T 8
anvi-run-kegg-kofams -c contigs.db -T 8
# 3. Map reads
bowtie2-build contigs.fa contigs
bowtie2 -x contigs -1 R1.fastq -2 R2.fastq -S sample.sam -p 8
samtools view -bS sample.sam | samtools sort -o sample.bam
anvi-init-bam sample.bam -o sample_sorted.bam
# 4. Profile
anvi-profile -i sample_sorted.bam -c contigs.db -o PROFILE
# 5. Binning (automatic)
anvi-cluster-contigs -p PROFILE/PROFILE.db -c contigs.db \
-C CONCOCT --driver CONCOCT -T 8
# 6. Interactive visualization
anvi-interactive -p PROFILE/PROFILE.db -c contigs.db
Anvi’o for Pangenomics
# Create genomes storage
anvi-gen-genomes-storage -e external-genomes.txt -o GENOMES.db
# Run pangenome
anvi-pan-genome -g GENOMES.db \
--project-name my_pangenome \
--num-threads 8
# Visualize
anvi-display-pan -g GENOMES.db -p my_pangenome/my_pangenome-PAN.db
Anvi’o Strengths
✅ Beautiful interactive interface
✅ Real-time binning (click to bin!)
✅ Metabolic reconstruction
✅ Pangenomics integration
✅ Publication-quality figures
🐍 4. ATLAS - Scalable Metagenomics
ATLAS uses Snakemake for large-scale analyses.
Installation
conda create -n atlas -c bioconda -c conda-forge metagenome-atlas
conda activate atlas
atlas init --db-dir databases
atlas download --db-dir databases
Running ATLAS
# Initialize project
atlas init --db-dir ~/databases my_project
# Edit config.yaml and samples.tsv
# Run complete workflow
atlas run all \
--working-dir my_project \
--config-file my_project/config.yaml \
--cores 32 \
--jobs 5
# Or specific steps
atlas run qc # Quality control
atlas run assembly # Assembly
atlas run binning # Binning
atlas run genomes # Genome refinement
ATLAS Features
✅ Automatic database downloads
✅ Co-assembly support
✅ Metatranscriptomics (RNA-seq integration)
✅ DRAM annotation
✅ Easy configuration
🧩 5. SqueezeMeta - Integrated Pipeline
SqueezeMeta = Complete pipeline with co-assembly.
Installation
git clone https://github.com/jtamames/SqueezeMeta.git
cd SqueezeMeta
./install_linux.sh
Running SqueezeMeta
# Single sample
SqueezeMeta.pl \
-m sequential \
-p project_name \
-s samples.txt \
-f raw_reads \
-t 24
# Co-assembly mode
SqueezeMeta.pl \
-m coassembly \
-p coassembly_project \
-s samples.txt \
-f raw_reads \
-t 24 \
--assembly megahit
SqueezeMeta Outputs
- Taxonomic classification (multiple databases)
- Functional annotation (KEGG, COG, Pfam)
- Metabolic pathways
- Interactive HTML reports
- Abundance tables
🌐 Part II: Web-Based Platforms
Why Use Web Platforms?
✅ No installation (works in browser)
✅ Pre-configured (databases ready)
✅ User-friendly (GUI interface)
✅ Collaborative (share projects easily)
✅ HPC access (use their servers)
❌ Limited customization
❌ Data upload time (can be slow)
❌ Storage limits (usually)
❌ Privacy concerns (your data on their servers)
🌌 1. Galaxy - Web-Based Analysis
Galaxy = Most popular web platform for bioinformatics
Getting Started
Public servers:
- Main: https://usegalaxy.org
- Europe: https://usegalaxy.eu
- Australia: https://usegalaxy.org.au
Create account → Free!
Galaxy Metagenomics Workflow
Step 1: Upload Data
- Click “Upload Data”
- Choose files or paste URLs
- Select “fastqsanger.gz” format
- Click “Start”
Step 2: Quality Control
Tool: FastQC
Tools → FASTQ Quality Control → FastQC
- Input: Your FASTQ files
- Execute
Tool: Trim Galore!
Tools → FASTQ Quality Control → Trim Galore!
- Single-end or Paired-end: Paired-end
- Reads 1: R1.fastq
- Reads 2: R2.fastq
- Execute
Step 3: Assembly
Tool: MEGAHIT
Tools → Assembly → MEGAHIT
- Single/Paired-end: Paired
- Forward: trimmed_R1.fastq
- Reverse: trimmed_R2.fastq
- Minimum contig length: 1500
- Execute
Step 4: Binning
Tool: MetaBAT2
Tools → Metagenomics → MetaBAT2
- Assembly: contigs.fasta
- BAM files: mapped_reads.bam
- Minimum contig length: 2500
- Execute
Step 5: Quality Check
Tool: CheckM
Tools → Metagenomics → CheckM lineage_wf
- Bins: MetaBAT2 bins
- Execute
Step 6: Annotation
Tool: Prokka
Tools → Annotation → Prokka
- For each bin
- Kingdom: Bacteria
- Execute
Galaxy Workflows
Create reusable workflows:
- Go to Workflow menu
- Click “Extract Workflow”
- Select history
- Name workflow
- Save
Run on new data:
- Upload new files
- Go to Workflows
- Select your workflow
- Map inputs
- Run!
Galaxy Advantages
✅ No installation
✅ 1000+ tools available
✅ Workflows can be shared
✅ Educational tutorials
✅ Free compute resources
🧬 2. KBase - Systems Biology Platform
KBase = DOE Joint Genome Institute platform
Website: https://kbase.us
Getting Started
- Create account (free)
- Create narrative (like Jupyter notebook)
- Upload data
- Add analysis apps
KBase Metagenomics Apps
Available Apps:
- Trim Reads (Trimmomatic)
- Assemble Reads (MEGAHIT, SPAdes)
- Annotate Assembly (RAST, PROKKA)
- Bin Contigs (MaxBin2, MetaBAT2)
- Classify Taxonomy (GTDB-Tk)
- Compare Genomes (Pangenome)
- Build Metabolic Model (ModelSEED)
Example KBase Workflow
1. Import FASTQ Data
App: "Import FASTQ File as Reads"
2. Quality Control
App: "Assess Read Quality with FastQC"
3. Trim Reads
App: "Trim Reads with Trimmomatic"
4. Assemble
App: "Assemble Reads with MEGAHIT"
5. Annotate
App: "Annotate Assembly with PROKKA"
6. Bin
App: "Bin Contigs with MetaBAT2"
7. Check Quality
App: "Assess Genome Quality with CheckM"
8. Classify
App: "Classify Genomes with GTDB-Tk"
9. Build Model
App: "Build Metabolic Model with ModelSEED"
KBase Strengths
✅ Integrated platform (data + analysis + models)
✅ Metabolic modeling (unique feature!)
✅ Reproducible narratives
✅ Collaborative
✅ DOE compute resources
KBase Limitations
❌ Steeper learning curve
❌ Fewer tools than Galaxy
❌ US-focused (slower internationally)
🗄️ 3. IMG/M - Integrated Microbial Genomes & Microbiomes
IMG/M = JGI’s comprehensive database + analysis platform
Website: https://img.jgi.doe.gov
What is IMG/M?
Not just analysis - It’s a massive database!
- 300,000+ bacterial genomes
- 40,000+ metagenomes
- Pre-computed annotations
- Comparative tools
Using IMG/M
Upload Your Data
- Create account (free)
- Submit metagenome
- Wait for processing (days-weeks)
- Receive annotated results
Or Browse Public Data
Search by:
- Ecosystem
- Taxonomy
- Genes
- Functions
- Pathways
IMG/M Features
Automatic annotation includes:
- COG/KOG categories
- Pfam domains
- KEGG pathways
- Enzyme functions
- Signal peptides
- Transmembrane helices
Comparative tools:
- Gene phylogenetic profiler
- Abundance profiles
- Function comparisons
- BLAST searches
IMG/M Best For
✅ Comparing your data to public datasets
✅ Exploring gene families
✅ Metabolic pathway analysis
✅ Publication-quality annotations
❌ Not for quick turnaround
❌ Can’t customize pipeline
🦠 4. BV-BRC (formerly PATRIC) - Bacterial & Viral Database
BV-BRC = Bacterial and Viral Bioinformatics Resource Center
Website: https://www.bv-brc.org
BV-BRC Services
Genome Assembly
Services → Assembly
- Upload reads
- Choose assembler
- Submit job
Genome Annotation
Services → Annotation
- Upload genome
- Select taxonomy
- Choose annotation options
- Submit
Comprehensive Genome Analysis
Services → Comprehensive Genome Analysis
- Upload reads OR genome
- One-stop analysis:
- Assembly
- Annotation
- Quality assessment
- Specialized analyses
BV-BRC Analysis Tools
- Proteome Comparison - Compare protein families
- Phylogenetic Tree - Build trees from genomes
- Protein Family Sorter - Find conserved proteins
- Comparative Pathway Analysis - KEGG pathway comparison
- Variation Analysis - SNP/indel identification
- RNA-Seq Analysis - Transcriptomics
- Tn-Seq Analysis - Transposon sequencing
BV-BRC Strengths
✅ Bacterial/viral focus
✅ AMR gene database (critical for pathogens)
✅ Drug target identification
✅ Extensive genome collection
✅ NIH-funded (reliable)
Best For
- Clinical/pathogen studies
- AMR research
- Vaccine development
- Outbreak analysis
🎯 Part III: Choosing the Right Tool
Decision Matrix
| Your Situation | Best Choice |
|---|---|
| New to bioinformatics | Galaxy |
| Need complete automation | MetaWRAP or nf-core/mag |
| Want interactive binning | Anvi’o |
| Large dataset (100+ samples) | ATLAS or nf-core/mag |
| Need metabolic modeling | KBase |
| Comparing to public data | IMG/M |
| Clinical/pathogen focus | BV-BRC |
| Publication-quality analysis | Anvi’o or nf-core/mag |
| Teaching/learning | Galaxy |
| Collaborative project | KBase or Galaxy |
💡 Best Practices
For Workflow Wrappers
- Start with default parameters
- Don’t over-customize initially
- Understand baseline before tweaking
-
Use version control
git init my_project git add config.yaml git commit -m "Initial config" - Document your choices
- Why this assembler?
- Why these QC thresholds?
- Keep a lab notebook 6
- Test on subset
- Don’t run 100 samples immediately
- Test 1-3 samples first
- Monitor resource usage
htop # Watch CPU/memory df -h # Check disk space
For Web Platforms
- Start with tutorials
- Galaxy has excellent training materials
- KBase has guided narratives
- Save workflows
- Create reusable workflows
- Share with collaborators
- Check data limits
- Know upload size limits
- Understand storage quotas
- Export results
- Download key files locally
- Don’t rely solely on web storage
- Cite properly
- Each platform has citation requirements
- Acknowledge compute resources
🎓 Learning Resources
MetaWRAP
nf-core/mag
Anvi’o
Galaxy
KBase
✅ Success Checklist
- Chose appropriate workflow wrapper OR web platform
- Successfully ran test dataset
- Understood output structure
- Validated results against manual analysis (Days 1-7)
- Documented workflow/parameters
- Saved/exported results
- Ready to scale up to full dataset
📚 Key Takeaways
- Workflow wrappers save time - Automate Days 1-7
- Web platforms lower barriers - No Linux/HPC needed
- Choose based on needs - No one-size-fits-all
- Start simple - Use defaults before customizing
- Reproducibility matters - Document everything
Repo for today’s code and other details
🔗 Day 8 →
Last updated: February 2026