Day 4 — Scaling Analysis: For Loops vs SLURM Job Arrays
Day 4: Scaling Analysis — For Loops vs SLURM Job Arrays
This is Day 4 of a 6-part series: From Laptop to HPC: Scaling Computational Biology Workflows and 🧬 Day 61 of Daily Bioinformatics from Jojy’s Desk. In Day 3 we learned how to submit a single job. Today we scale it: the same work, 200× faster.
The Problem With Sequential Processing
You’ve written your SLURM script. You know how to run one job. Now your supervisor hands you 200 samples and says the analysis needs to be done by Friday.
Your first instinct is a for loop:
for sample in data/*.fastq.gz; do
sbatch process_one_sample.sh "$sample"
done
Or maybe a single job with a loop inside:
#!/bin/bash
#SBATCH --time=50:00:00
for sample in data/*.fastq.gz; do
fastqc "$sample" -o qc_results/
done
Both of these work. But they’re leaving enormous potential on the table.
The for loop approach submits 200 separate jobs manually. The single-job-with-loop approach runs everything sequentially — sample 2 waits for sample 1 to finish, sample 3 waits for sample 2, and so on.
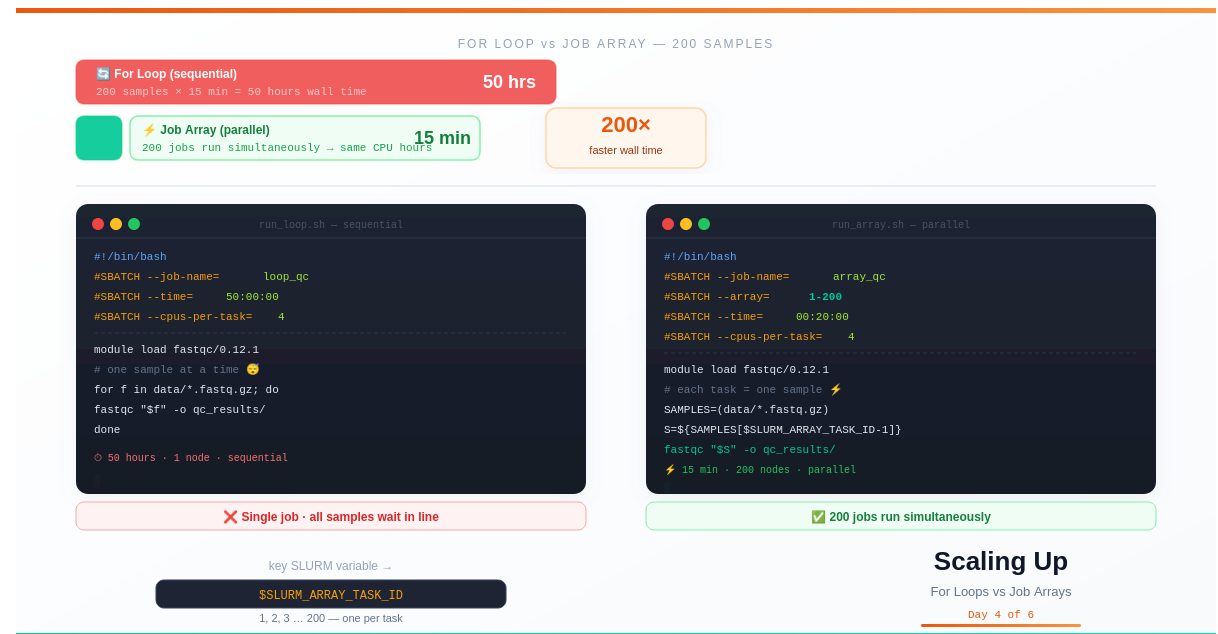
With 200 samples at 15 minutes each, you’re looking at 50 hours of wall time.
Job arrays run all 200 simultaneously. Wall time: 15 minutes.
Same total CPU hours. Completely different calendar time.
What You’ll Learn Today
- Why sequential processing is the wrong default for large datasets
- What a SLURM job array actually is and how it works
- How
$SLURM_ARRAY_TASK_IDmaps to your sample files - How to control concurrency — running 20 at a time instead of all 200
- How to handle failed tasks, rerun subsets, and read per-task logs
- Real patterns: paired-end reads, sample sheets, parameter sweeps
- A benchmark comparison: for loop vs job array
Part 1: What Is a Job Array?
A SLURM job array is a single sbatch submission that creates multiple identical jobs, each identified by a unique task ID — an integer from 1 to N.
Every task runs the same script, but gets a different task ID through the environment variable $SLURM_ARRAY_TASK_ID. Your script uses that ID to figure out which sample it should process.
Think of it like this:
sbatch --array=1-200 process.sh
↓
SLURM creates 200 jobs:
Job 84600_1 → process.sh (SLURM_ARRAY_TASK_ID=1)
Job 84600_2 → process.sh (SLURM_ARRAY_TASK_ID=2)
Job 84600_3 → process.sh (SLURM_ARRAY_TASK_ID=3)
...
Job 84600_200 → process.sh (SLURM_ARRAY_TASK_ID=200)
All 200 run in parallel across available nodes.
The array job ID (84600) is shared across all tasks. Each task has its own task ID (1–200). Together they form a unique identifier: 84600_1, 84600_2, etc.
Part 2: Your First Job Array
Let’s convert a for loop into a job array step by step.
Starting Point: The For Loop
#!/bin/bash
# run_loop.sh — processes samples sequentially
module load fastqc/0.12.1
mkdir -p qc_results/
for sample in data/*.fastq.gz; do
fastqc "$sample" -o qc_results/
done
# ⏱ 200 samples × 15 min = 50 hours wall time
Step 1: Add the #SBATCH –array directive
#!/bin/bash
#SBATCH --job-name=fastqc_array
#SBATCH --array=1-200 # create 200 tasks
#SBATCH --output=logs/%A_%a.out # %A=array ID, %a=task ID
#SBATCH --error=logs/%A_%a.err
#SBATCH --time=00:30:00 # per-task time limit
#SBATCH --mem=4G # per-task memory
#SBATCH --cpus-per-task=4
module purge
module load fastqc/0.12.1
mkdir -p qc_results/ logs/
# Step 2: Use $SLURM_ARRAY_TASK_ID to pick one sample
SAMPLES=(data/*.fastq.gz) # bash array of all files
SAMPLE=${SAMPLES[$SLURM_ARRAY_TASK_ID - 1]} # pick the Nth one
echo "Task $SLURM_ARRAY_TASK_ID processing: $SAMPLE"
fastqc "$SAMPLE" \
-t $SLURM_CPUS_PER_TASK \
-o qc_results/
echo "Done: $(date)"
# ⚡ 200 tasks run in parallel → 15 min wall time
Submit it:
sbatch run_array.sh
# Submitted batch job 84600
That one command launches 200 jobs. All of them start as soon as compute nodes become available.
What Does $SLURM_ARRAY_TASK_ID - 1 Mean?
Bash arrays are 0-indexed (first element is index 0), but SLURM arrays start at 1 by default. Subtracting 1 aligns them:
SLURM_ARRAY_TASK_ID=1 → index 0 → data/sample_001.fastq.gz
SLURM_ARRAY_TASK_ID=2 → index 1 → data/sample_002.fastq.gz
SLURM_ARRAY_TASK_ID=3 → index 2 → data/sample_003.fastq.gz
...
SLURM_ARRAY_TASK_ID=200 → index 199 → data/sample_200.fastq.gz
If you prefer to start your array at 0:
#SBATCH --array=0-199
SAMPLE=${SAMPLES[$SLURM_ARRAY_TASK_ID]} # no subtraction needed
Part 3: Monitoring Job Arrays
# See all your array tasks
squeue -u $USER
# The output shows each task separately:
# JOBID PARTITION NAME USER ST TIME NODES
# 84600_1 normal fastqc_array you R 0:03 1
# 84600_2 normal fastqc_array you R 0:03 1
# 84600_[3-200]normal fastqc_array you PD 0:00 1
# Compact view (groups pending tasks in brackets)
squeue -u $USER -t all
# Check one specific task
scontrol show job 84600_42
# Get efficiency for one task
seff 84600_42
Reading Per-Task Log Files
Because you used %A_%a in your output filename, every task has its own log:
logs/84600_1.out # task 1
logs/84600_2.out # task 2
...
logs/84600_200.out # task 200
# Check if any tasks failed
grep -l "error\|Error\|FAILED" logs/84600_*.err
# Check a specific task's output
cat logs/84600_42.out
# See how many tasks completed successfully
grep -l "Done:" logs/84600_*.out | wc -l
Part 4: Controlling Concurrency — The %N Throttle
Sometimes you don’t want all 200 tasks running at once. Reasons include:
- The cluster has limited nodes and you don’t want to monopolise them
- A shared database or reference file has read limits
- You want to be a polite cluster citizen
Control maximum simultaneous tasks with %N:
# Run at most 20 tasks at a time
#SBATCH --array=1-200%20
# Run at most 50 tasks at a time
#SBATCH --array=1-200%50
# Run them all (default)
#SBATCH --array=1-200
With --array=1-200%20, SLURM starts 20 tasks, and as each one finishes, it immediately starts the next pending one. The queue self-manages — you don’t need to babysit it.
Part 5: Rerunning Failed Tasks
In real workflows, some tasks fail. Maybe a sample had corrupted data. Maybe a node went down mid-job. Job arrays make it trivial to rerun only the failures.
Finding Which Tasks Failed
# Check exit codes for all tasks
sacct -j 84600 --format=JobID,State,ExitCode
# Output:
# 84600_1 COMPLETED 0:0
# 84600_2 COMPLETED 0:0
# 84600_5 FAILED 1:0 ← this one failed
# 84600_42 FAILED 1:0 ← and this one
# ...
# Quick one-liner to list just the failed task IDs
sacct -j 84600 --format=JobID,State -n | \
awk '$2=="FAILED" {print $1}' | \
sed 's/84600_//'
# outputs: 5, 42
Rerunning Specific Tasks
# Rerun only tasks 5 and 42
#SBATCH --array=5,42
# Rerun a range of tasks
#SBATCH --array=10-20
# Combine: specific tasks plus a range
#SBATCH --array=5,42,100-110
Or cancel a specific task that’s running but producing bad output:
# Cancel one task
scancel 84600_42
# Cancel all pending tasks in the array (let running ones finish)
scancel -t PENDING 84600
# Cancel the entire array
scancel 84600
Part 6: Real Bioinformatics Patterns
Pattern 1: Paired-End FASTQ Files
Most sequencing data comes in pairs (R1 and R2). Your array needs to handle both files for each sample.
#!/bin/bash
#SBATCH --array=1-100
#SBATCH --job-name=align_pe
#SBATCH --output=logs/%A_%a.out
#SBATCH --cpus-per-task=8
#SBATCH --mem=16G
#SBATCH --time=02:00:00
module purge
module load bowtie2/2.5.1 samtools/1.17
# Build arrays of R1 and R2 files
R1_FILES=(data/*_R1.fastq.gz)
R2_FILES=(data/*_R2.fastq.gz)
R1=${R1_FILES[$SLURM_ARRAY_TASK_ID - 1]}
R2=${R2_FILES[$SLURM_ARRAY_TASK_ID - 1]}
# Derive sample name from R1 filename
SAMPLE=$(basename "$R1" _R1.fastq.gz)
echo "Processing sample: $SAMPLE"
echo "R1: $R1"
echo "R2: $R2"
mkdir -p alignments/
bowtie2 \
-x ref/genome \
-1 "$R1" \
-2 "$R2" \
-p $SLURM_CPUS_PER_TASK \
2> alignments/${SAMPLE}.log | \
samtools sort \
-@ $SLURM_CPUS_PER_TASK \
-o alignments/${SAMPLE}.bam
samtools index alignments/${SAMPLE}.bam
echo "Alignment complete: $(date)"
Pattern 2: Sample Sheet (CSV)
For complex projects, maintain a CSV sample sheet instead of relying on filename patterns:
sample_id,r1_path,r2_path,condition
ctrl_01,data/ctrl_01_R1.fastq.gz,data/ctrl_01_R2.fastq.gz,control
ctrl_02,data/ctrl_02_R1.fastq.gz,data/ctrl_02_R2.fastq.gz,control
treat_01,data/treat_01_R1.fastq.gz,data/treat_01_R2.fastq.gz,treatment
#!/bin/bash
#SBATCH --array=2-51 # rows 2-51 of the CSV (row 1 = header)
#SBATCH --job-name=align_sheet
#SBATCH --output=logs/%A_%a.out
#SBATCH --cpus-per-task=8
#SBATCH --mem=16G
#SBATCH --time=02:00:00
module purge
module load bowtie2/2.5.1 samtools/1.17
SHEET="samples.csv"
# Extract this task's row from the CSV
ROW=$(awk -F',' "NR==$SLURM_ARRAY_TASK_ID" "$SHEET")
SAMPLE=$(echo "$ROW" | cut -d',' -f1)
R1=$(echo "$ROW" | cut -d',' -f2)
R2=$(echo "$ROW" | cut -d',' -f3)
COND=$(echo "$ROW" | cut -d',' -f4)
echo "Sample: $SAMPLE | Condition: $COND"
mkdir -p alignments/
bowtie2 -x ref/genome -1 "$R1" -2 "$R2" \
-p $SLURM_CPUS_PER_TASK \
| samtools sort -@ $SLURM_CPUS_PER_TASK \
-o alignments/${SAMPLE}.bam
samtools index alignments/${SAMPLE}.bam
Pattern 3: Parameter Sweep
Job arrays aren’t just for processing different samples. They’re perfect for testing a range of parameters:
#!/bin/bash
#SBATCH --array=1-10
#SBATCH --job-name=param_sweep
#SBATCH --output=logs/sweep_%A_%a.out
#SBATCH --cpus-per-task=4
#SBATCH --mem=8G
module load kraken2/2.1.3
# Test 10 different confidence thresholds (0.1 to 1.0)
CONFIDENCE=$(echo "scale=1; $SLURM_ARRAY_TASK_ID / 10" | bc)
echo "Testing confidence threshold: $CONFIDENCE"
mkdir -p results/sweep_${CONFIDENCE}/
kraken2 \
--db /scratch/databases/kraken2_standard \
--confidence "$CONFIDENCE" \
--report results/sweep_${CONFIDENCE}/report.txt \
data/test_sample.fastq.gz \
> results/sweep_${CONFIDENCE}/output.txt
echo "Confidence $CONFIDENCE done: $(date)"
Part 7: The Full Benchmark
Here’s the comparison across all three approaches for 200 samples, 15 minutes processing time each:
| Approach | Wall Time | CPU Hours | Notes |
|---|---|---|---|
| Laptop for loop | ~50 hours | 50 | Sequential, laptop tied up |
| Single HPC job with loop | ~50 hours | 50 | Better resources, still sequential |
| HPC job array (200 tasks) | ~15 minutes | 50 | All tasks run simultaneously |
| HPC job array (throttled 50%) | ~30 minutes | 50 | Max 100 tasks at once |
The total CPU hours are identical in every case — you’re not doing less work. The difference is pure parallelism. Job arrays don’t reduce compute; they compress wall time.
This matters enormously in practice:
- A 50-hour sequential job must finish without interruption
- If it crashes at hour 48, you lose everything and restart
- A 15-minute array job is easy to rerun; each task is independent
- Failures affect one task, not the whole analysis
Part 8: Common Mistakes and How to Fix Them
Array size doesn’t match number of files
# ❌ If you have 200 files but array=1-199
# Task 200 is never run — one sample missed silently
# ✅ Count your files first
ls data/*.fastq.gz | wc -l
# 200
#SBATCH --array=1-200 # matches exactly
Off-by-one errors
# ❌ SLURM starts at 1, bash arrays at 0
SAMPLES=(data/*.fastq.gz)
SAMPLE=${SAMPLES[$SLURM_ARRAY_TASK_ID]} # task 1 → index 1 (skips sample 1)
# ✅ Always subtract 1 when using --array=1-N
SAMPLE=${SAMPLES[$SLURM_ARRAY_TASK_ID - 1]} # task 1 → index 0 (correct)
Requesting too many resources per task
# ❌ Overkill — wastes queue priority
#SBATCH --mem=64G # for a FastQC job that uses 2 GB
# ✅ Profile one sample first with an interactive node
# then set realistic limits
#SBATCH --mem=4G
Not checking for silent failures
# ❌ Job shows COMPLETED but output is wrong
# FastQC might fail on a corrupted file but exit 0
# ✅ Add explicit checks
if [[ ! -f "qc_results/${SAMPLE}_fastqc.html" ]]; then
echo "ERROR: output not created for $SAMPLE"
exit 1
fi
Forgetting to create output and log directories
# ❌ Job fails: cannot open output file
#SBATCH --output=logs/%A_%a.out
# → logs/ doesn't exist
# ✅ Create directories in your pre-submission step
mkdir -p logs/ qc_results/ alignments/
sbatch run_array.sh
Try It Yourself
# 1. Create 10 test files
mkdir -p test_array/data/ test_array/logs/
cd test_array/
# Generate 10 small fake FASTQ files for testing
for i in $(seq -w 1 10); do
echo -e "@read1\nACGT\n+\nIIII" > data/sample_${i}.fastq
done
# 2. Write the array script
cat > run_array.sh << 'EOF'
#!/bin/bash
#SBATCH --job-name=test_array
#SBATCH --array=1-10
#SBATCH --output=logs/%A_%a.out
#SBATCH --time=00:05:00
#SBATCH --mem=500M
#SBATCH --cpus-per-task=1
SAMPLES=(data/*.fastq)
SAMPLE=${SAMPLES[$SLURM_ARRAY_TASK_ID - 1]}
echo "Task $SLURM_ARRAY_TASK_ID → $SAMPLE"
echo "Lines in file: $(wc -l < $SAMPLE)"
sleep 10
echo "Done: $(date)"
EOF
# 3. Submit
sbatch run_array.sh
# 4. Watch all 10 tasks run simultaneously
watch squeue -u $USER
# 5. Check outputs
ls logs/
grep "Task" logs/*.out
Summary
- For loops are sequential — simple to write but all samples wait in line
- Job arrays run one task per sample, all in parallel — same total compute, fraction of the wall time
-
$SLURM_ARRAY_TASK_IDis the key: each task uses it to select its own input file - Use
%A_%ain output filenames for per-task logs (array job ID + task ID) - Throttle with
--array=1-200%50to limit concurrent tasks - Rerun only failed tasks by specifying their IDs:
--array=5,42 - 200 samples × 15 min = 50 hours sequential → 15 minutes parallel — that’s the single biggest win in HPC bioinformatics
Up Next
Day 5: Reproducible Pipelines — Snakemake and Nextflow
You can now run any analysis on HPC at scale. But shell scripts and job arrays have a hidden problem: they don’t track dependencies. If step 2 depends on step 1 finishing, and step 1 fails halfway through, your script doesn’t know. In Day 5, we introduce workflow managers that solve this — and make your science reproducible.