Building Custom HUMAnN3 Databases with Struo2 and GTDB: A Realistic Guide from the Trenches
Building Custom HUMAnN3 Databases with Struo2 and GTDB r220: A Realistic Guide from the Trenches
Introduction
Creating custom HUMAnN3 databases sounds straightforward in theory, but in practice, it’s a journey filled with environment conflicts, memory issues, and mysterious errors. This guide documents the real process of building a custom HUMAnN3 database using Struo2 with GTDB release 220, including all the troubleshooting, workarounds, and hard-won lessons.
Spoiler alert: If you need results quickly, download the pre-built databases from Struo2’s FTP server. But if you want to learn the process or need a truly custom database, read on. The pre built database is built on an older version of GTDBtk, thats why I tried to build newone.
Why Build Custom Databases?
- Latest taxonomy: GTDB r220 (released 2024) has significantly improved archaeal and bacterial classifications
- Ecosystem-specific: Focus on genomes relevant to your study system
- Updated gene annotations: Latest UniRef annotations for functional profiling
- Research reproducibility: Control exactly which genomes are included
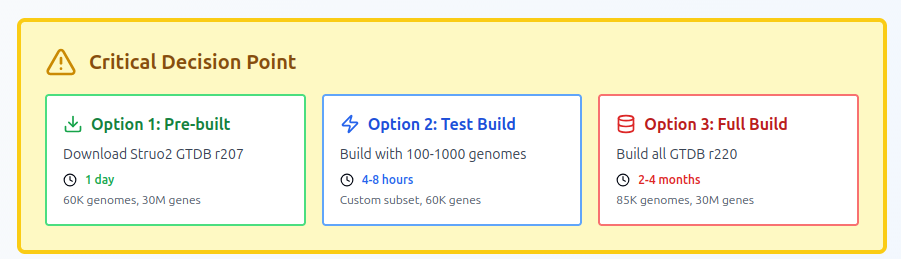
Reality Check: Resource Requirements
Minimal Setup (100 test genomes)
- Time: 4-8 hours
- RAM: 200-250 GB
- CPUs: 32 cores
- Storage: 500 GB
- Outcome: Proof of concept, ~60K genes
Full GTDB r220 Setup (~85,000 genomes)
- Time: 2-4 months (yes, months!)
- RAM: 500+ GB
- CPUs: 90+ cores recommended
- Storage: 10+ TB
- Outcome: Comprehensive database, ~30 million genes
My recommendation: Start with 100-1000 representative genomes unless you have unlimited compute time. I used this to fix all run erorr and in the mean time I was looking to find a quick solution.
Step 1: Environment Setup (The First Battle)
The PATH Problem
The biggest challenge isn’t Struo2 itself—it’s environment conflicts. Here’s what you’ll encounter:
# You think you're using conda environment A
conda activate struo2_fresh
# But your PATH says otherwise
which python
# /some/old/environment/bin/python ← NOT what you want!
Solution: Clean your environment completely:
# Deactivate EVERYTHING
conda deactivate
conda deactivate
conda deactivate
# Check your .bashrc for sneaky PATH additions
grep -n "export PATH" ~/.bashrc
# Comment out any conda auto-init or old environment additions
nano ~/.bashrc
# Start fresh
source ~/.bashrc
conda activate struo2_fresh
Create a Clean Snakemake Environment
Don’t mix Struo2 dependencies with Snakemake—keep them separate:
# Snakemake environment (just for running the pipeline)
conda create -n snakemake_env python=3.9 -y
conda activate snakemake_env
conda install -c conda-forge -c bioconda snakemake=7.32.4 -y
# Load system modules instead of installing everything via conda
module load vsearch/2.21.1
module load bowtie2/2.4.4
module load samtools/1.15.1
module load diamond/2.0.13
module load seqkit/2.10.0
Critical: DO NOT use --use-conda with Snakemake if your HPC has SSL certificate issues!
Step 2: Download GTDB Data
Get the Metadata
mkdir -p data/taxdump_r220
cd data
# Download GTDB r220 metadata
wget https://data.gtdb.ecogenomic.org/releases/release220/220.0/ar53_metadata_r220.tsv
wget https://data.gtdb.ecogenomic.org/releases/release220/220.0/bac120_metadata_r220.tsv
# Download taxonomy files
wget http://ftp.tue.mpg.de/ebio/projects/struo2/GTDB_release220/taxdump/names.dmp -P taxdump_r220/
wget http://ftp.tue.mpg.de/ebio/projects/struo2/GTDB_release220/taxdump/nodes.dmp -P taxdump_r220/
Download Genomes (The Smart Way)
Option 1: Test subset (100 genomes)
# Create a test table with 100 diverse genomes
head -101 ar53_metadata_r220.tsv | tail -100 > gtdb-r220_test100.tsv
# Download genomes using GTDB accessions
# Use ncbi-genome-download or direct GTDB downloads
Option 2: Download pre-built Struo2 database (RECOMMENDED)
# Much faster than building from scratch!
git clone https://github.com/leylabmpi/Struo2.git
cd Struo2
# Download GTDB r207 complete database (~60K genomes)
./util_scripts/database_download.py -t 8 -r 207 \
-d humann3 metadata taxdump -- ../gtdb_r207_prebuilt
# This downloads everything in ~1 day vs 2-4 months building
Download UniRef Database
mkdir -p data/mmseqs2_dbs/uniref90
# Download UniRef90 (for gene annotation)
mmseqs databases UniRef90 data/mmseqs2_dbs/uniref90/uniref90 data/mmseqs2_TMP --remove-tmp-files 1
Step 3: Prepare Your Sample Table
Create samples_single_per_species.tsv:
ncbi_organism_name accession fasta_file_path gtdb_taxid gtdb_taxonomy
Methanosarcina_mazei RS_GCF_000970205.1 /path/to/genome.fna.gz 3005035806 d__Archaea;p__Halobacteriota;...
Critical tip: Use ONE genome per species to avoid Pandas DataFrame bugs in Struo2:
# If you have multiple assemblies per species
awk 'NR==1 || !seen[$1]++' samples_full.tsv > samples_single_per_species.tsv
Step 4: Configure Struo2
Create config_custom.yaml:
samples_file: /path/to/samples_single_per_species.tsv
output_dir: ./databases/
tmp_dir: ./tmp/
databases:
kraken2: Skip
bracken: Skip
genes: Create
humann3_bowtie2: Create
humann3_diamond: Create
uniref_name: uniref90
dmnd_name: uniref90_201901.dmnd
names_dmp: ./data/taxdump_r220/names.dmp
nodes_dmp: ./data/taxdump_r220/nodes.dmp
keep_intermediate: True
params:
humann3:
batches: 4
mmseqs_search:
db: ./data/mmseqs2_dbs/uniref90/uniref90
index: -s 6
run: -e 1e-3 --max-accept 1 --max-seqs 100
Step 5: The MMseqs Problem (CPU Architecture Incompatibility)
Problem you WILL encounter:
Illegal instruction (core dumped)
Error: indexdb died
Why: The system module for MMseqs was compiled for a different CPU architecture.
Solution: Download a compatible MMseqs binary:
cd ~
wget https://mmseqs.com/latest/mmseqs-linux-avx2.tar.gz
tar xvzf mmseqs-linux-avx2.tar.gz
export PATH=$HOME/mmseqs/bin:$PATH
# Test it works
mmseqs version
mmseqs createdb --help # Should NOT crash
Step 6: Create the SLURM Script
Here’s a production-ready script that handles all the gotchas:
#!/bin/bash
#SBATCH --job-name=struo2_humann3
#SBATCH --time=72:00:00
#SBATCH --cpus-per-task=32
#SBATCH --mem=250G
#SBATCH --output=struo2_%j.log
#SBATCH --error=struo2_%j.err
cd /path/to/struo2_fresh
# Critical: Use downloaded MMseqs, not module
export PATH=$HOME/mmseqs/bin:$PATH
# Remove any conflicting paths
export PATH=$(echo $PATH | tr ':' '\n' | grep -v "metawrap\|metaWRAP" | tr '\n' ':')
# SSL certificates (if needed)
export REQUESTS_CA_BUNDLE=/etc/ssl/certs/ca-bundle.crt
export SSL_CERT_FILE=/etc/ssl/certs/ca-bundle.crt
export CURL_CA_BUNDLE=/etc/ssl/certs/ca-bundle.crt
source ~/.bashrc
conda activate snakemake_env
# Load modules
module load vsearch/2.21.1
module load bowtie2/2.4.4
module load samtools/1.15.1
module load diamond/2.0.13
module load seqkit/2.10.0
echo "Verifying tools:"
echo " mmseqs: $(which mmseqs)"
mmseqs version
# Run WITHOUT --use-conda to avoid SSL/conda issues
snakemake \
-j 32 \
--configfile config_custom.yaml \
--keep-going \
--rerun-incomplete \
--printshellcmds
echo "Completed: $(date)"
Step 7: Common Errors and Solutions
Error 1: SSL Certificate Issues
RuntimeError: Download error (77) Problem with the SSL CA cert
Solution: DON’T use --use-conda. Load system modules instead.
Error 2: Bowtie2 --threads Not Recognized
bowtie2-build: unrecognized option '--threads'
Your cluster has old Bowtie2 (v2.2.3). Build indices manually:
cd databases/humann3/uniref90/
gunzip -c genome_reps_filt_annot.fna.gz > temp.fna
bowtie2-build temp.fna genome_reps_filt_annot # No --threads flag!
rm temp.fna
Error 3: Out of Memory with Bowtie2
Out of memory allocating the offs[] array for the Bowtie index
Why: 30 million genes = ~180GB Bowtie2 index
Solution: Request 300GB RAM minimum
Error 4: Wrong Database Version
CRITICAL ERROR: ChocoPhlAn contains files that are not of expected version
Solution: Add --bypass-nucleotide-index to HUMAnN3 commands
Step 8: Running HUMAnN3 with Your Database
Single Sample Test
#!/bin/bash
#SBATCH --cpus-per-task=16
#SBATCH --mem=300G
#SBATCH --time=24:00:00
module load humann/3.9
NUC_DB=/path/to/databases/humann3/uniref90
PROT_DB=/path/to/databases/protein
humann --input sample.fasta \
--output output_dir \
--nucleotide-database $NUC_DB \
--protein-database $PROT_DB \
--bypass-nucleotide-index \
--bypass-prescreen \
--threads 16
Batch Processing (32 samples)
#!/bin/bash
#SBATCH --job-name=humann3_batch
#SBATCH --array=1-32
#SBATCH --cpus-per-task=16
#SBATCH --mem=300G
#SBATCH --time=48:00:00
module load humann/3.9
SAMPLES=($(ls /path/to/assemblies/*.fasta))
SAMPLE=${SAMPLES[$SLURM_ARRAY_TASK_ID-1]}
BASENAME=$(basename $SAMPLE .fasta)
humann --input $SAMPLE \
--output /path/to/output \
--nucleotide-database $NUC_DB \
--protein-database $PROT_DB \
--output-basename ${BASENAME} \
--bypass-nucleotide-index \
--bypass-prescreen \
--threads 16
Step 9: Understanding Your Output
What You Get
databases/
├── genes/
│ ├── genome_reps_filtered.fna.gz # 22 MB (100 genomes)
│ ├── genome_reps_filtered.faa.gz # 16 MB
│ └── genome_reps_filtered.txt.gz # 2 MB
└── humann3/
├── uniref90/
│ ├── genome_reps_filt_annot.fna.gz # 25 GB (full GTDB)
│ ├── genome_reps_filt_annot.tsv.gz # 3.3 GB - THE KEY FILE!
│ └── *.bt2l # ~180 GB total
└── protein/
└── uniref90.dmnd # 36 GB
The Magic TSV File
genome_reps_filt_annot.tsv.gz contains gene-to-genome mappings:
seq_uuid genome_name species annotation
hash123 RS_GCF_000970205.1 s__Methanosarcina_mazei UniRef90_Q8TUR2
This file lets you:
- Map gene families back to genomes
- Calculate genome-level abundances
- Build trait tables for functional redundancy analysis
Lessons Learned
What Worked
✅ Separate Snakemake environment from tool dependencies
✅ System modules instead of conda for tools
✅ Downloaded MMseqs binary instead of system module
✅ One genome per species to avoid Pandas bugs
✅ Starting with 100 genomes for testing
What Didn’t Work
❌ Using --use-conda with SSL certificate issues
❌ Mixing conda environments
❌ System MMseqs module (CPU incompatibility)
❌ Building Bowtie2 index with old version’s flags
❌ Trying to build full GTDB without months of compute time
Time Investment
- Setup and troubleshooting: 2-3 days
- Test run (100 genomes): 4-8 hours
- Full build (if attempted): 2-4 months
- Using pre-built database: 1 day
Alternative Strategies
Strategy 1: Smart Subsetting (This is the way I find to proceed)
Instead of using all 85K genomes:
- Run HUMAnN3 on your samples with pre-built database (older gtdbtk based)
- Extract list of detected genomes (from gene mappings)
- Download only those ~500-5000 genomes
- Build custom database from your ecosystem-specific subset
- Re-run samples for accurate genome abundances
This is probably the best approach for most projects!
Strategy 2: Use MAGs based custom database
If you alredy have MAGs from same data, you can use them instead of the genomes from GTDBtk. But remebr that you still have the issue of limited coverage, as MAGs many not give you all genomes in each samples.
Final Recommendations
If you want to analyze data TODAY:
- Download pre-built Struo2 GTDB r207 database
- Use MetaPhlAn for taxonomy
- Use HUMAnN3 for functional profiling
If you have 1 week:
- Build custom database with ~1000 representative genomes
- Test thoroughly with pilot samples
- Scale to full dataset
If you have 2-4 months and unlimited compute:
- Build full GTDB r220 database
- Enjoy the most comprehensive reference available
- Share it with the community!
Computational Costs
For our HPC cluster (Clemson University Palmetto):
100 genome test:
- SUs consumed: ~500
- Cost: ~$50
- Time: 8 hours
Full GTDB build (estimated):
- SUs consumed: ~50,000+
- Cost: ~$5,000+
- Time: 2-4 months
Using pre-built database:
- SUs consumed: ~10 (download only)
- Cost: ~$1
- Time: 1 day
Conclusions
Building custom HUMAnN3 databases with Struo2 is powerful but challenging. The pipeline works, but you’ll fight with:
- Environment conflicts
- Memory requirements
- CPU architecture incompatibilities
- Time constraints
For most projects, I recommend:
- Use pre-built databases for initial analysis
- Identify genomes relevant to your system
- Build targeted custom databases if needed
The functional profiling is worth it—just be realistic about the time investment!
Resources
- Struo2 GitHub: https://github.com/leylabmpi/Struo2
- Pre-built databases: http://ftp.tue.mpg.de/ebio/projects/struo2/
- GTDB: https://gtdb.ecogenomic.org/
- HUMAnN3: https://huttenhower.sph.harvard.edu/humann/
Special thanks to Claude for helping troubleshoot every single error message in this journey!