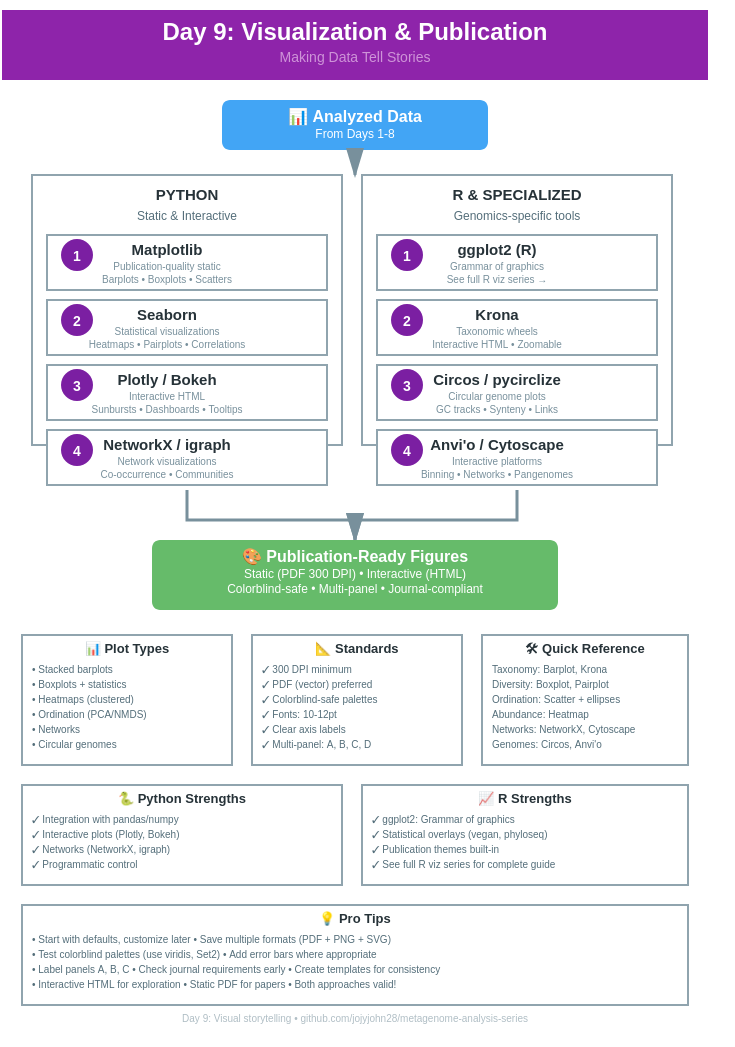
Day 9: Visualization & Publication - Making Data Tell Stories
| Estimated Time: 4-6 hours | Difficulty: Intermediate | Prerequisites: Days 1-8 (Analyzed data ready) |
📚 Table of Contents
- Introduction
- Part I: Quick R/ggplot2 Overview
- Part II: Python Visualizations
- Part III: Specialized Tools
- Part IV: Interactive Visualizations
- Publication Guidelines
🎯 Introduction
You’ve generated amazing data. Now make it beautiful.
Why Visualization Matters
Bad visualization:
- ❌ Hides patterns
- ❌ Confuses readers
- ❌ Gets papers rejected
Good visualization:
- ✅ Reveals patterns instantly
- ✅ Tells compelling stories
- ✅ Makes papers memorable
Today’s Goal
Create publication-quality figures that journals accept and readers love.
📊 Part I: Quick R/ggplot2 Overview
ggplot2 Basics
Quick example:
library(ggplot2)
# Abundance plot
ggplot(abundance_data, aes(x = Sample, y = Abundance, fill = Phylum)) +
geom_bar(stat = "identity") +
theme_minimal() +
labs(title = "Community Composition",
x = "Sample", y = "Relative Abundance (%)") +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
Key ggplot2 Plots for Metagenomics
- Stacked barplots - Taxonomic composition
- Boxplots - Alpha diversity
- PCA/NMDS - Ordination
- Heatmaps - Gene/MAG abundance
- Faceted plots - Multiple samples
Try ggplots and compare your image with toy data plots. look for R in the figure name.
🐍 Part II: Python Visualizations
Why Python for Visualization?
✅ Matplotlib - Publication-quality static plots
✅ Seaborn - Statistical visualizations
✅ Plotly - Interactive plots
✅ Bokeh - Dashboard-style plots
✅ Integration - Works with pandas, numpy
📊 1. Matplotlib - Foundation
Installation
pip install matplotlib seaborn pandas numpy
Personal advice : Use visual code studio for all plots.
Basic Taxonomy Barplot
Try Basic taxonomy barplot and compare your image with basic_taxonomy_py
Alpha Diversity Boxplot
Try Alpha Diversity Boxplot and compare your image with alpha_diversity_py
🎨 2. Seaborn - Statistical Plots
Heatmap with Clustering
Try Heatmap with Clustering and compare your image with heatmap
Correlation Heatmap
Try correlation and compare your image with Correlation_py
Pairplot for Multivariate Data
Try Pairplot and compare your image with Pairplot
🌐 3. Plotly - Interactive Plots
Interactive Taxonomy Sunburst
Try Interactive taxonomy .py and compare your image with interactive tax plot
Interactive Scatter with Annotations
Try Scatter plot and compare your image with scatter-toydata
See my full visualization series for:
👉 Heatmaps
🍩 Part III: Specialized Tools
🌳 1. Krona - Taxonomic Visualization
Krona creates beautiful interactive taxonomic wheels.
Installation
# Download Krona
git clone https://github.com/marbl/Krona.git
cd Krona/KronaTools
./install.pl
# Update taxonomy
./updateTaxonomy.sh
Creating Krona Plot
# From Kraken2 output
ktImportTaxonomy \
-q 2 -t 3 \
kraken2_output.txt \
-o krona_taxonomy.html
# From BLAST results
ktImportBLAST \
blast_results.txt \
-o krona_blast.html
# From taxonomy table
# Format: count, taxonomy (tab-separated)
ktImportText \
taxonomy_counts.txt \
-o krona_custom.html
Krona Input Format
# taxonomy_counts.txt
150 Bacteria;Proteobacteria;Gammaproteobacteria;Enterobacterales
89 Bacteria;Firmicutes;Bacilli;Lactobacillales
67 Bacteria;Bacteroidetes;Bacteroidia;Bacteroidales
45 Bacteria;Actinobacteria;Actinobacteria;Actinomycetales
Output: Interactive HTML with zoom, search, and hierarchy navigation
⭕ 2. Circos - Genome Comparisons
Circos creates circular genome visualizations.
Installation
# Install via conda
conda install -c bioconda circos
# Or download
wget http://circos.ca/distribution/circos-0.69-9.tgz
tar -xzf circos-0.69-9.tgz
cd circos-0.69-9
Basic Circos Plot
# circos.conf
karyotype = karyotype.txt
<ideogram>
<spacing>
default = 0.005r
</spacing>
radius = 0.90r
thickness = 20p
fill = yes
</ideogram>
<plots>
# GC content
<plot>
type = heatmap
file = gc_content.txt
r1 = 0.88r
r0 = 0.83r
color = blues-9-seq
</plot>
# Gene density
<plot>
type = histogram
file = gene_density.txt
r1 = 0.82r
r0 = 0.77r
fill_color = green
</plot>
</plots>
<links>
<link>
file = genome_comparisons.txt
radius = 0.75r
color = red_a4
thickness = 2
</link>
</links>
<<include etc/colors_fonts_patterns.conf>>
<<include etc/housekeeping.conf>>
Running Circos
circos -conf circos.conf -outputdir output -outputfile genome_circos.png
Python Alternative: pycirclize
from pycirclize import Circos
import pandas as pd
# Initialize
circos = Circos(sectors={"chr1": 10, "chr2": 15, "chr3": 12})
# Add tracks
for sector in circos.sectors:
track = sector.add_track((80, 100))
track.axis()
# GC content
track.heatmap(gc_data)
# Gene positions
track.xticks_by_interval(1000000)
# Add links between genomes
circos.link(("chr1", 1000, 2000), ("chr2", 5000, 6000))
# Save
circos.savefig("genome_comparison.pdf")
🕸️ 3. Network Visualizations
igraph in Python
import igraph as ig
import matplotlib.pyplot as plt
import pandas as pd
# Load co-occurrence network
edges = pd.read_csv('cooccurrence_network.csv')
# Create graph
g = ig.Graph.TupleList(edges.itertuples(index=False),
directed=False,
edge_attrs=['weight'])
# Calculate metrics
g.vs['degree'] = g.degree()
g.vs['betweenness'] = g.betweenness()
# Community detection
communities = g.community_multilevel()
g.vs['community'] = communities.membership
# Visual style
visual_style = {
'vertex_size': [d*3 for d in g.vs['degree']],
'vertex_color': g.vs['community'],
'vertex_label': g.vs['name'],
'edge_width': [w*2 for w in g.es['weight']],
'layout': g.layout_fruchterman_reingold(),
'bbox': (800, 800),
'margin': 50
}
# Plot
ig.plot(g, 'network.pdf', **visual_style)
NetworkX + Matplotlib
import networkx as nx
import matplotlib.pyplot as plt
import pandas as pd
# Create network
G = nx.Graph()
# Add edges from co-occurrence data
edges = pd.read_csv('cooccurrence_network.csv')
for _, row in edges.iterrows():
G.add_edge(row['MAG1'], row['MAG2'], weight=row['correlation'])
# Remove weak connections
G_filtered = G.copy()
edges_to_remove = [(u, v) for u, v, d in G_filtered.edges(data=True)
if abs(d['weight']) < 0.3]
G_filtered.remove_edges_from(edges_to_remove)
# Layout
pos = nx.spring_layout(G_filtered, k=0.5, iterations=50)
# Node sizes by degree
node_sizes = [G_filtered.degree(node) * 100 for node in G_filtered.nodes()]
# Node colors by community
communities = nx.community.greedy_modularity_communities(G_filtered)
node_colors = []
for node in G_filtered.nodes():
for i, comm in enumerate(communities):
if node in comm:
node_colors.append(i)
break
# Plot
plt.figure(figsize=(14, 14))
# Draw edges
edges = G_filtered.edges()
weights = [G_filtered[u][v]['weight'] for u, v in edges]
nx.draw_networkx_edges(G_filtered, pos, alpha=0.3, width=weights)
# Draw nodes
nx.draw_networkx_nodes(G_filtered, pos,
node_size=node_sizes,
node_color=node_colors,
cmap=plt.cm.Set3,
alpha=0.8)
# Draw labels (only for large nodes)
large_nodes = [node for node in G_filtered.nodes()
if G_filtered.degree(node) > 5]
labels = {node: node for node in large_nodes}
nx.draw_networkx_labels(G_filtered, pos, labels, font_size=8)
plt.title('MAG Co-occurrence Network', fontsize=16, fontweight='bold')
plt.axis('off')
plt.tight_layout()
plt.savefig('cooccurrence_network.pdf', dpi=300, bbox_inches='tight')
plt.show()
Cytoscape Export
# Export for Cytoscape
import pandas as pd
# Nodes file
nodes = pd.DataFrame({
'id': list(G.nodes()),
'degree': [G.degree(n) for n in G.nodes()],
'betweenness': [nx.betweenness_centrality(G)[n] for n in G.nodes()]
})
nodes.to_csv('network_nodes.csv', index=False)
# Edges file
edges_export = []
for u, v, d in G.edges(data=True):
edges_export.append({
'source': u,
'target': v,
'weight': d['weight']
})
edges_df = pd.DataFrame(edges_export)
edges_df.to_csv('network_edges.csv', index=False)
print("Import these files into Cytoscape for advanced styling")
🎨 4. Advanced Heatmaps
ComplexHeatmap Style in Python
import matplotlib.pyplot as plt
import seaborn as sns
import pandas as pd
import numpy as np
from scipy.cluster import hierarchy
from scipy.spatial.distance import pdist
# Load data
abundance = pd.read_csv('mag_abundance.csv', index_col=0)
metadata = pd.read_csv('metadata.csv', index_col=0)
# Log transform
abundance_log = np.log10(abundance + 1)
# Hierarchical clustering
row_linkage = hierarchy.linkage(pdist(abundance_log), method='ward')
col_linkage = hierarchy.linkage(pdist(abundance_log.T), method='ward')
# Create figure with GridSpec
fig = plt.figure(figsize=(14, 12))
gs = fig.add_gridspec(3, 3,
height_ratios=[0.3, 3, 0.2],
width_ratios=[0.2, 3, 0.3],
hspace=0.05, wspace=0.05)
# Top annotation (metadata)
ax_top = fig.add_subplot(gs[0, 1])
treatment_colors = {'Control': '#3498db', 'Treatment': '#e74c3c'}
treatment_bars = [treatment_colors[metadata.loc[s, 'Treatment']]
for s in abundance.columns]
ax_top.imshow([treatment_bars], aspect='auto')
ax_top.set_xticks([])
ax_top.set_yticks([0])
ax_top.set_yticklabels(['Treatment'])
ax_top.spines['top'].set_visible(False)
ax_top.spines['right'].set_visible(False)
# Left dendrogram
ax_left = fig.add_subplot(gs[1, 0])
hierarchy.dendrogram(row_linkage, orientation='left', ax=ax_left)
ax_left.set_xticks([])
ax_left.set_yticks([])
ax_left.spines['top'].set_visible(False)
ax_left.spines['right'].set_visible(False)
ax_left.spines['bottom'].set_visible(False)
ax_left.spines['left'].set_visible(False)
# Main heatmap
ax_main = fig.add_subplot(gs[1, 1])
im = ax_main.imshow(abundance_log, aspect='auto', cmap='viridis')
ax_main.set_xticks(range(len(abundance.columns)))
ax_main.set_xticklabels(abundance.columns, rotation=90)
ax_main.set_yticks(range(len(abundance.index)))
ax_main.set_yticklabels(abundance.index)
# Colorbar
ax_cbar = fig.add_subplot(gs[1, 2])
plt.colorbar(im, cax=ax_cbar, label='log10(Abundance + 1)')
plt.savefig('complex_heatmap.pdf', dpi=300, bbox_inches='tight')
plt.show()
🔬 Part IV: Interactive Visualizations
1. Anvi’o Interactive Interface
Already covered in Day 8, but key features:
# Interactive binning
anvi-interactive -p PROFILE.db -c CONTIGS.db
# Interactive pangenome
anvi-display-pan -g GENOMES.db -p PAN.db
# Features:
# - Real-time binning
# - Metadata integration
# - Beautiful circular displays
# - Export publication figures
2. Bokeh Dashboard
from bokeh.plotting import figure, show, output_file
from bokeh.models import HoverTool, ColumnDataSource
from bokeh.layouts import column, row
import pandas as pd
# Load data
abundance = pd.read_csv('mag_abundance.csv')
# Create ColumnDataSource
source = ColumnDataSource(data=dict(
x=abundance['Sample'],
y=abundance['MAG_1'],
desc=abundance['Taxonomy']
))
# Create figure
p = figure(x_range=abundance['Sample'].unique(),
height=400, width=800,
title="MAG Abundance Across Samples",
toolbar_location="above")
# Add bars
p.vbar(x='x', top='y', width=0.8, source=source,
color='navy', alpha=0.8)
# Add hover tool
hover = HoverTool(tooltips=[
("Sample", "@x"),
("Abundance", "@y"),
("Taxonomy", "@desc")
])
p.add_tools(hover)
# Styling
p.xaxis.major_label_orientation = 45
p.xgrid.grid_line_color = None
# Save
output_file("interactive_abundance.html")
show(p)
📐 Publication Guidelines
General Principles
- Resolution: 300 DPI minimum
- File formats:
- PDF (vector, preferred)
- PNG (high-res raster)
- TIFF (some journals require)
- Dimensions: Check journal requirements
- Colors: Colorblind-friendly palettes
- Fonts: Arial, Helvetica, or Times (10-12pt)
Color Palettes (Colorblind-Safe)
# Colorblind-friendly palettes
import matplotlib.pyplot as plt
# Qualitative (categorical data)
cb_friendly_qual = ['#E69F00', '#56B4E9', '#009E73', '#F0E442',
'#0072B2', '#D55E00', '#CC79A7']
# Sequential (continuous data)
cb_friendly_seq = plt.cm.viridis # or 'plasma', 'cividis'
# Diverging (centered data)
cb_friendly_div = plt.cm.RdBu_r
# Use in plots
plt.scatter(x, y, c=categories, cmap=cb_friendly_qual)
Figure Checklist
- High resolution (300 DPI)
- Proper file format (PDF/PNG)
- Clear axis labels with units
- Readable font sizes (≥10pt)
- Colorblind-friendly colors
- Legend clearly labeled
- No overlapping text
- Consistent style across panels
- Error bars where appropriate
- Statistical annotations added
Multi-Panel Figures
import matplotlib.pyplot as plt
from matplotlib.gridspec import GridSpec
# Create figure
fig = plt.figure(figsize=(16, 10))
gs = GridSpec(2, 3, figure=fig, hspace=0.3, wspace=0.3)
# Panel A: Taxonomy barplot
ax1 = fig.add_subplot(gs[0, :2])
# ... plot code ...
ax1.text(-0.1, 1.05, 'A', transform=ax1.transAxes,
fontsize=16, fontweight='bold', va='top')
# Panel B: Alpha diversity
ax2 = fig.add_subplot(gs[0, 2])
# ... plot code ...
ax2.text(-0.15, 1.05, 'B', transform=ax2.transAxes,
fontsize=16, fontweight='bold', va='top')
# Panel C: Ordination
ax3 = fig.add_subplot(gs[1, 0])
# ... plot code ...
ax3.text(-0.15, 1.05, 'C', transform=ax3.transAxes,
fontsize=16, fontweight='bold', va='top')
# Panel D: Heatmap
ax4 = fig.add_subplot(gs[1, 1:])
# ... plot code ...
ax4.text(-0.08, 1.05, 'D', transform=ax4.transAxes,
fontsize=16, fontweight='bold', va='top')
plt.savefig('figure1_composite.pdf', dpi=300, bbox_inches='tight')
plt.show()
💡 Best Practices
1. Data Preparation
# Always clean data first
def prepare_data_for_plotting(df):
# Remove zeros
df = df[(df > 0).any(axis=1)]
# Log transform
df_log = np.log10(df + 1)
# Normalize if needed
df_norm = df.div(df.sum(axis=0), axis=1) * 100
return df_log, df_norm
2. Consistent Styling
# Set global matplotlib style
import matplotlib.pyplot as plt
plt.rcParams.update({
'font.size': 12,
'font.family': 'sans-serif',
'font.sans-serif': ['Arial'],
'axes.linewidth': 1.5,
'axes.labelsize': 12,
'axes.titlesize': 14,
'xtick.labelsize': 10,
'ytick.labelsize': 10,
'legend.fontsize': 10,
'figure.dpi': 100,
'savefig.dpi': 300,
'savefig.bbox': 'tight'
})
3. Save Multiple Formats
def save_figure(fig, name):
"""Save figure in multiple formats"""
fig.savefig(f'{name}.pdf', dpi=300, bbox_inches='tight')
fig.savefig(f'{name}.png', dpi=300, bbox_inches='tight')
fig.savefig(f'{name}.svg', bbox_inches='tight') # For editing
print(f"✓ Saved {name} in PDF, PNG, and SVG formats")
✅ Success Checklist
- Created taxonomic composition plots
- Generated alpha diversity figures
- Made ordination plots (PCA/NMDS)
- Created heatmaps (abundance, correlation)
- Built network visualizations
- Generated interactive HTML plots
- Used Krona for taxonomy
- All figures are publication-quality (300 DPI)
- Used colorblind-friendly palettes
- Saved in multiple formats
📚 Key Resources
Python:
Specialized:
Repo for today’s code and other details
🔗 Day 9
Last updated: February 2026