Day 2 — Clean Your RNA : QC & Preprocessing for Metatranscriptomics
Here is a truth most tutorials skip: the quality of your metatranscriptome analysis is decided before you ever run a mapper.
Downstream tools — Bowtie2, featureCounts, DESeq2 — are sophisticated, but they cannot rescue bad input data. Low-quality reads reduce alignment rates. Adapter contamination generates spurious mappings. Unremoved rRNA floods your count table with noise and buries the mRNA signal you actually care about. A pipeline that begins with dirty RNA produces a dirty result, even if every subsequent step is executed perfectly.
Garbage RNA in → garbage expression out.
This is the step that separates careful work from fast work. Let’s do it carefully.
Why RNA data is especially messy
RNA-seq data has all the quality issues of DNA sequencing, plus several of its own:
Adapter contamination. Libraries are built by ligating adapter sequences onto your cDNA fragments. If your insert is shorter than the read length — common with degraded RNA — the sequencer reads straight into the adapter. These adapter sequences are not biological and will either fail to map or map to the wrong place.
Low-quality 3′ bases. Sequencing quality degrades toward the end of reads. Phred scores below Q20 mean more than 1 in 100 bases is wrong. For short environmental RNA reads this degrades alignment substantially.
rRNA dominance. Even with ribosomal depletion kits, 60–95% of reads in a metatranscriptome are ribosomal RNA. The ribosome is the most transcribed molecular machine in any living cell — this is unavoidable biology, not a kit failure. These reads must be removed computationally before mapping.
Paired-end inconsistency. After trimming, some read pairs lose one mate (one mate trimmed below the minimum length threshold). These orphaned reads need to be tracked or excluded — most downstream tools expect matched pairs, and mismatched files cause silent errors.
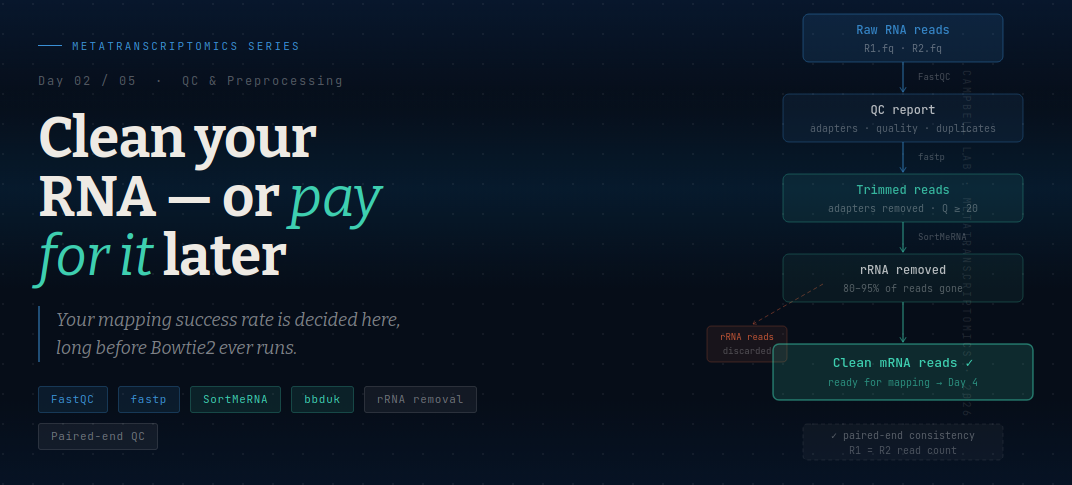
The preprocessing pipeline
The full workflow has four stages, run in this order:
Raw reads → FastQC (diagnose) → fastp (trim) → SortMeRNA (remove rRNA) → clean mRNA reads
Step 1 — FastQC: know what you have before touching it
Before trimming anything, run FastQC. It takes minutes and prevents hours of debugging.
module load fastqc
fastqc \
CP_Spr15G08_R1.paired.fq \
CP_Spr15G08_R2.paired.fq \
-o fastqc_raw/ \
-t 8
Open the HTML report and check five things:
| Module | What to look for |
|---|---|
| Per base sequence quality | Median Phred ≥ Q28 across the read; a 3′ drop is normal |
| Adapter content | Any Illumina adapter flagged = you must trim |
| Per sequence quality scores | Distribution should peak above Q30 |
| Sequence duplication levels | >50% warrants attention in metatranscriptomics |
| Per base N content | Should be near zero; spikes = flow cell problem |
FastQC diagnoses only — it does not fix anything. Run it before trimming and again after, to confirm your preprocessing worked.
Step 2 — fastp: trim adapters and low-quality bases
fastp is the preferred trimmer for metatranscriptomics. It handles paired-end reads gracefully, detects adapters automatically, and produces an HTML report alongside the cleaned reads.
module load fastp
fastp \
-i CP_Spr15G08_R1.paired.fq \
-I CP_Spr15G08_R2.paired.fq \
-o CP_Spr15G08_R1.trimmed.fq \
-O CP_Spr15G08_R2.trimmed.fq \
--unpaired1 CP_Spr15G08_unpaired.fq \
--unpaired2 CP_Spr15G08_unpaired.fq \
--detect_adapter_for_pe \
--qualified_quality_phred 20 \
--length_required 50 \
--thread 8 \
--html CP_Spr15G08_fastp.html \
--json CP_Spr15G08_fastp.json
Flag breakdown:
-
--detect_adapter_for_pe— auto-detects adapters in paired-end mode; no need to specify sequences manually -
--qualified_quality_phred 20— removes reads where average quality drops below Q20 -
--length_required 50— discards reads shorter than 50 bp after trimming (too short to map reliably) -
--unpaired1 / --unpaired2— saves orphaned reads separately rather than discarding them silently -
--html / --json— produces a quality report you should always check
Alternative — Trimmomatic: If you need to exactly replicate parameters from a published study, Trimmomatic gives finer control with sliding-window trimming:
trimmomatic PE \
CP_Spr15G08_R1.paired.fq \
CP_Spr15G08_R2.paired.fq \
R1_paired.fq R1_unpaired.fq \
R2_paired.fq R2_unpaired.fq \
ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 \
LEADING:3 TRAILING:3 \
SLIDINGWINDOW:4:20 \
MINLEN:50
For most metatranscriptomics workflows, fastp defaults are sufficient and considerably faster.
Step 3 — SortMeRNA: remove ribosomal RNA
This is the most important preprocessing step specific to metatranscriptomics. SortMeRNA compares your reads against curated rRNA databases (Silva, Rfam) using a k-mer index and separates rRNA reads from putative mRNA reads.
module load sortmerna
sortmerna \
--ref $SILVA/silva-bac-16s-id90.fasta \
--ref $SILVA/silva-arc-16s-id95.fasta \
--ref $SILVA/silva-euk-18s-id95.fasta \
--ref $SILVA/rfam-5s-database-id98.fasta \
--reads CP_Spr15G08_R1.trimmed.fq \
--reads CP_Spr15G08_R2.trimmed.fq \
--aligned rRNA_reads/CP_Spr15G08_rRNA \
--other mRNA_reads/CP_Spr15G08_mRNA \
--fastx \
--paired-in \
--out2 \
--threads 8 \
--workdir sortmerna_tmp/CP_Spr15G08
Key flags:
-
--ref— pass each rRNA database separately; use all four for comprehensive coverage (bacterial 16S, archaeal 16S, eukaryotic 18S, 5S/5.8S) -
--aligned— destination for rRNA reads (you keep these for QC checks but don’t map them) -
--other— your mRNA-enriched output; this is what you carry forward -
--paired-in— treats both mates as rRNA if either mate matches a database entry -
--out2— writes separate R1 and R2 output files (required for paired-end downstream tools) -
--workdir— SortMeRNA writes a large KV store here; use a separate directory per sample
How much rRNA should you expect?
| Depletion protocol | Typical rRNA fraction |
|---|---|
| No depletion (total RNA) | 90–97% |
| Commercial depletion kit | 50–80% |
| Poly-A selection (eukaryote-biased) | 5–20% |
| After SortMeRNA removal | <5% remaining |
After SortMeRNA, expect your read count to drop to roughly 5–40% of the pre-removal total depending on how effective your wet-lab depletion was. This is not read loss — this is noise removal.
Alternative — bbduk (BBTools): Faster than SortMeRNA, slightly less sensitive for divergent sequences. Excellent when processing many samples in parallel:
bbduk.sh \
in1=CP_Spr15G08_R1.trimmed.fq \
in2=CP_Spr15G08_R2.trimmed.fq \
out1=CP_Spr15G08_R1.mRNA.fq \
out2=CP_Spr15G08_R2.mRNA.fq \
ref=ribokmers.fa.gz \
k=31 \
ordered=t \
threads=8
The ribokmers.fa.gz file ships with BBTools. For well-characterized systems bbduk and SortMeRNA perform comparably; for novel environments with divergent rRNA, SortMeRNA with multiple databases is more thorough.
Step 4 — Post-trimming FastQC: verify your work
Always run FastQC again on the mRNA-enriched reads. A successful preprocessing run should show:
- ✅ No adapter content flags
- ✅ Per-base quality stable across read length (the 3′ drop is gone)
- ✅ Read length distribution consistent with your trimming parameters
- ✅ R1 and R2 have identical read counts (paired-end consistency)
fastqc \
CP_Spr15G08_R1.mRNA.fq \
CP_Spr15G08_R2.mRNA.fq \
-o fastqc_clean/ \
-t 8
If adapters still appear after fastp, or quality remains poor, something went wrong in trimming. Do not proceed to mapping with a failed QC. Diagnose first.
Checking your read counts at each stage
Track read counts through the pipeline to catch silent failures:
# Raw reads
echo "Raw R1:" $(cat CP_Spr15G08_R1.paired.fq | wc -l) / 4 | bc
# After fastp
echo "Trimmed R1:" $(cat CP_Spr15G08_R1.trimmed.fq | wc -l) / 4 | bc
# After SortMeRNA
echo "mRNA R1:" $(cat mRNA_reads/CP_Spr15G08_mRNA_fwd.fq | wc -l) / 4 | bc
A typical progression for our estuarine samples:
Raw: 3,647,405 pairs
Trimmed: 3,512,000 pairs (~96% retained — good)
mRNA: 420,000 pairs (~12% of raw — expected with depletion kit)
The large drop at the rRNA removal step is expected and healthy. If your mRNA fraction is above 60% after SortMeRNA, double-check that your depletion kit worked — you may be mapping mostly rRNA in downstream steps.
When Your Paired Reads Fall Out of Sync: Repair Scripts & Why They Matter
The silent killer of metatranscriptomics pipelines: mismatched R1/R2 read counts after preprocessing. What causes it, what it breaks downstream, and how to fix it
The problem nobody warns you about
You run FastQC. Reads look clean. You run fastp. You run SortMeRNA. You check the output folders — files are there. You move confidently to mapping and run Bowtie2.
Then this happens:
Error: reads file 2 contains a different number of reads
than reads file 1. Terminating Bowtie2.
Or with featureCounts, you get a count table full of zeros and no obvious error message. Or SAMtools flagstat returns 0 properly paired reads when you expected thousands.
Your R1 and R2 files have different numbers of reads. The paired-end contract is broken. And it happened silently somewhere in your preprocessing.
This is one of the most common and frustrating problems in metatranscriptomics pipelines — and it is completely fixable if you know to look for it.
What is paired-end sequencing and why does the count matter?
In paired-end RNA-seq, each cDNA fragment is sequenced from both ends. R1 reads the forward strand; R2 reads the reverse strand of the same fragment. They are linked — read number 1 in R1 corresponds exactly to read number 1 in R2. Read 500,000 in R1 corresponds to read 500,000 in R2.
This is not just a convention. Every paired-end tool — Bowtie2, STAR, featureCounts with -p, DESeq2 — assumes this positional correspondence is intact. When R1 has 420,000 reads and R2 has 418,347 reads, that assumption is violated everywhere from line 418,348 onward. The tools either crash with an error (the good outcome) or silently produce wrong results (the dangerous outcome).
# Quick check — these numbers must match
echo "R1 reads: $(cat mRNA_reads/CP_Spr15G08_mRNA_fwd.fq | wc -l | awk '{print $1/4}')"
echo "R2 reads: $(cat mRNA_reads/CP_Spr15G08_mRNA_rev.fq | wc -l | awk '{print $1/4}')"
If these numbers differ by even one read, you have a problem.
Why does this happen? The three main causes
Cause 1: SortMeRNA asymmetric filtering
This is the most common cause in metatranscriptomics pipelines specifically.
SortMeRNA decides per read whether it matches rRNA databases. With --paired-in, it flags a pair as rRNA if either mate matches. But in some versions and configurations, the output split between --aligned and --other can produce unequal mate counts — especially when reads are processed in chunks across threads, or when the --workdir KV store becomes corrupted mid-run.
When you run SortMeRNA without --out2, it writes a single interleaved output. When you split that output or pass it to tools expecting separate files, the interleaving can get broken. Always use --out2 and always check counts afterward.
Cause 2: fastp orphan handling
fastp removes reads that fall below quality or length thresholds after trimming. In paired-end mode, when one mate in a pair is discarded (because trimming left it shorter than --length_required), the surviving mate becomes an orphan — it has no partner.
If you write orphans to the same output files as paired reads (a common mistake), your R1 and R2 files end up with different counts. If you discard orphans entirely without tracking them, you silently lose data. The --unpaired1 and --unpaired2 flags in fastp exist exactly to prevent this — they route orphans to a separate file, keeping R1 and R2 synchronized.
Cause 3: Manual or pipeline file operations
Any grep, head, split, cat, or file copy applied to only one of your paired files can silently desynchronize them. This also happens when a job runs out of walltime mid-write — SLURM kills the job while R1 is being written, leaving R2 complete and R1 truncated. Always check file sizes and read counts after any operation that writes FASTQ files.
Why this matters so much for mapping
Bowtie2 maps paired-end reads by looking for concordant alignments — both mates mapping to the same reference contig, in the right orientation, within the expected insert size. This concordant signal is what gives you high-confidence mappings and the “properly paired” statistic in SAMtools flagstat.
When your files are out of sync, Bowtie2 cannot find concordant pairs — not because your reference is incomplete, but because the R1/R2 positional correspondence is broken. You’ll see:
0 (0.00%) aligned concordantly exactly 1 time
0 (0.00%) properly paired
This looks identical to a genuine reference coverage problem (like our 2% rate from Day 4). The difference is: a coverage problem is biological and expected. A pairing problem is a data integrity issue and will silently corrupt every downstream analysis — featureCounts, DESeq2, DNA:RNA ratios, all of it.
The consequence: you might think your MAGs simply aren’t expressed. In reality, your data is broken and any signal is noise.
The fix: two approaches
Option A — bbduk repair.sh (BBTools)
repair.sh is purpose-built for this exact problem. It reads both FASTQ files, matches read IDs between them, and outputs three files: synchronized R1, synchronized R2, and a singletons file for reads whose mate was lost entirely.
repair.sh \
in1=CP_Spr15G08_mRNA_fwd.fq \
in2=CP_Spr15G08_mRNA_rev.fq \
out1=CP_Spr15G08_R1.repaired.fq \
out2=CP_Spr15G08_R2.repaired.fq \
outs=CP_Spr15G08_singletons.fq \
repair=t \
threads=8 \
-Xmx28g
How it works: repair.sh loads all read IDs from both files into memory, identifies which IDs appear in one file but not the other, and writes only the matched pairs to output. Unmatched reads go to the singletons file — you keep them separately but do not use them in paired-end mapping.
Memory note: repair.sh loads read IDs into RAM. For large files (>50M reads per file), request at least 64 GB. The -Xmx flag sets the Java heap size — set it slightly below your --mem allocation.
When to use repair.sh: when you already have mismatched files and need to rescue them. It is a post-hoc fix.
Option B — fastp with strict paired-end enforcement
fastp can prevent the mismatch from ever occurring if run with the right flags. The key is ensuring that when one mate is discarded, the other is never written to the main output:
fastp \
-i CP_Spr15G08_R1.paired.fq \
-I CP_Spr15G08_R2.paired.fq \
-o CP_Spr15G08_R1.trimmed.fq \
-O CP_Spr15G08_R2.trimmed.fq \
--unpaired1 CP_Spr15G08_R1.unpaired.fq \
--unpaired2 CP_Spr15G08_R2.unpaired.fq \
--detect_adapter_for_pe \
--qualified_quality_phred 20 \
--length_required 50 \
--disable_adapter_trimming_for_paired \
--thread 8
The critical flags:
-
--unpaired1and--unpaired2route orphans to separate files — they never contaminate the paired output - fastp guarantees that R1 output and R2 output always have identical read counts when these flags are set
When to use fastp for this: prevention, not repair. Run it correctly the first time and you should never need repair.sh — unless SortMeRNA introduces the mismatch afterward.
The recommended workflow order
Raw reads
↓
fastp (with --unpaired1/2) ← prevents mismatch from trimming
↓
CHECK: R1 == R2 read count?
↓
SortMeRNA (with --out2) ← --out2 keeps pairs synchronized
↓
CHECK: R1 == R2 read count? ← if not equal, run repair.sh here
↓
repair.sh (if needed)
↓
Final CHECK: R1 == R2?
↓
→ Mapping (Day 4) — with confidence
Two check points: immediately after fastp, and immediately after SortMeRNA. If either check fails, run repair.sh before proceeding. Do not proceed to mapping with mismatched files.
Quick diagnostic: are my files in sync?
Add this function to your pipeline scripts:
check_pairs() {
local R1=$1
local R2=$2
local SAMPLE=$3
R1_N=$(cat "$R1" | wc -l | awk '{print $1/4}')
R2_N=$(cat "$R2" | wc -l | awk '{print $1/4}')
if [ "$R1_N" -eq "$R2_N" ]; then
echo "✓ $SAMPLE — paired: $R1_N reads each"
else
echo "✗ $SAMPLE — MISMATCH: R1=$R1_N R2=$R2_N — run repair.sh"
exit 1
fi
}
# Usage:
check_pairs mRNA_reads/CP_Spr15G08_mRNA_fwd.fq \
mRNA_reads/CP_Spr15G08_mRNA_rev.fq \
"CP_Spr15G08"
This function is embedded in all the SLURM scripts in the companion repository.
What about singletons — can I still use them?
Yes. Singletons (reads whose mate was lost) can be mapped as single-end reads and included in featureCounts. They contribute to gene counts, though without the concordance signal of proper pairs. Whether to include them is a judgment call:
- For quantification (TPM, DESeq2): include singletons — they add signal
- For structural analyses that depend on insert size: exclude them
- For this series: we focus on paired reads for clarity, but the SLURM scripts save singletons separately so you can use them if needed
# Map singletons separately as single-end reads
bowtie2 \
-x master_MAGs_index \
-U CP_Spr15G08_singletons.fq \
-p 8 \
-S CP_Spr15G08_singletons.sam
The takeaway
Paired-end read synchronization is a prerequisite for every downstream step. A single out-of-sync file can make a working pipeline produce results that are wrong in ways that are hard to detect — zeros where there should be signal, artificially low alignment rates, inflated variance in DESeq2.
The fix is simple: two read count checks and one repair script. Build them into your pipeline from the start and you will never diagnose this problem from the wrong end — hours into a mapping run, staring at a count table full of zeros.
→ Day 3: Reference Strategy — Your Database Defines Your Biology
Running all samples as a SLURM batch job
For three samples (or thirty), use the SLURM script provided in Day2_QC_Preprocessing/scripts/. See the companion repository for the full batch script.
📦 All code, SLURM scripts, and toy datasets used in this series are available in the companion repository 📦 Github repository
The takeaway
Preprocessing is not a formality. It is the foundation every downstream result is built on. The 30–45 minutes you spend on FastQC, fastp, and SortMeRNA protects every hour of analysis that follows — and explains a significant portion of the alignment rate you will see on coming days.
_Questions about this workflow? Drop a comment below. The complete R script and SLURM submission scripts are available in the companion GitHub repository.
If you seeing the blog first time, read DAY 1