Whole Genome Sequencing — Day 3: Taxonomy, Phylogeny & Genome Similarity
🧬 Whole Genome Sequencing — Day 3
Taxonomy, Phylogeny & Genome Similarity
In Day 1, we focused on raw read quality control. In Day 2, we assembled genomes and evaluated assembly quality and topology.
Today, in Day 3, we move from sequence reconstruction to biological identity.
👉Who is this genome related to? 👉Where does it sit in the tree of life? 👉Is it a known species or something new?
🎯 Goal of Day 3
Place genomes in an evolutionary and taxonomic context
Specifically, we aim to:
✔ Assign standardized genome-based taxonomy ✔ Construct phylogenomic trees ✔ Test species and genus boundaries ✔ Support NCBI submissions, MRAs, and comparative studies
This workflow is designed for dozens to hundreds of genomes, including:
• Isolate genomes
• MAG collections
• Mixed genome datasets
🧠 Why Genome-Based Taxonomy?
Historically, microbial taxonomy relied heavily on 16S rRNA genes. While useful, 16S alone:
• Lacks species-level resolution
• Is often fragmented or missing in MAGs
• Cannot resolve closely related taxa
Genome-based taxonomy solves these issues by using:
• Hundreds of conserved marker genes
• Whole-genome similarity metrics
• Curated, standardized reference frameworks
This is now the recommended approach for modern microbial genomics.
🧪 Core Analyses in Day 3
1️⃣ Genome Taxonomy Assignment
Assign taxonomy using conserved marker genes rather than single loci.
2️⃣ Phylogenomic Tree Construction
Infer evolutionary relationships using genome-scale alignments.
3️⃣ Species & Genus Boundary Testing
Evaluate novelty and relatedness using:
• ANI (nucleotide-level)
• AAI (protein-level)
• dDDH (digital DNA–DNA hybridization)
Using multiple metrics together provides robust taxonomic conclusions.
🧰 Tools Used
• GTDB-Tk – Genome-based taxonomy and phylogeny
• iTOL – Interactive phylogenomic tree visualization
• FastANI – Species-level genome similarity
• CompareM – Amino acid identity across genomes
• GGDC – Digital DNA–DNA hybridization (web-based)
• NCBI Datasets CLI – Reference genome retrieval
Each tool contributes a different layer of confidence.
🧬 GTDB-Tk: Genome-Based Taxonomy
GTDB-Tk (Genome Taxonomy Database Toolkit) assigns taxonomy based on a curated set of conserved marker genes and reference phylogenies.
1️⃣ Installing GTDB-Tk (Conda)
conda create -n gtdbtk_env -c bioconda -c conda-forge gtdbtk
conda activate gtdbtk_env
gtdbtk --version
For large projects, installing in a custom path is recommended due to database size.
2️⃣ Downloading the GTDB Reference Database
GTDB-Tk requires the full GTDB reference database (>100 GB).
wget https://data.gtdb.ecogenomic.org/releases/release214/214.1/gtdbtk/gtdbtk_r214_data.tar.gz
tar -xzf gtdbtk_r214_data.tar.gz -C /project/bcampb7/camplab/databases/
Set the environment variable:
export GTDBTK_DATA_PATH=/project/bcampb7/camplab/databases/gtdbtk_r214_data
echo "export GTDBTK_DATA_PATH=/project/bcampb7/camplab/databases/gtdbtk_r214_data" >> ~/.bashrc
gtdbtk check_install
3️⃣ GTDB-Tk Standard Workflow
(Identify → Align → Classify)
Identify conserved marker genes
gtdbtk identify \
--genome_dir genomes/ \
--out_dir gtdbtk/identify \
--extension fa \
--cpus 32
Align marker genes
gtdbtk align \
--identify_dir gtdbtk/identify \
--out_dir gtdbtk/align \
--cpus 32
Classify genomes
gtdbtk classify \
--genome_dir genomes/ \
--align_dir gtdbtk/align \
--out_dir gtdbtk/classify \
--cpus 32 \
--mash_db gtdbtk/mash_db
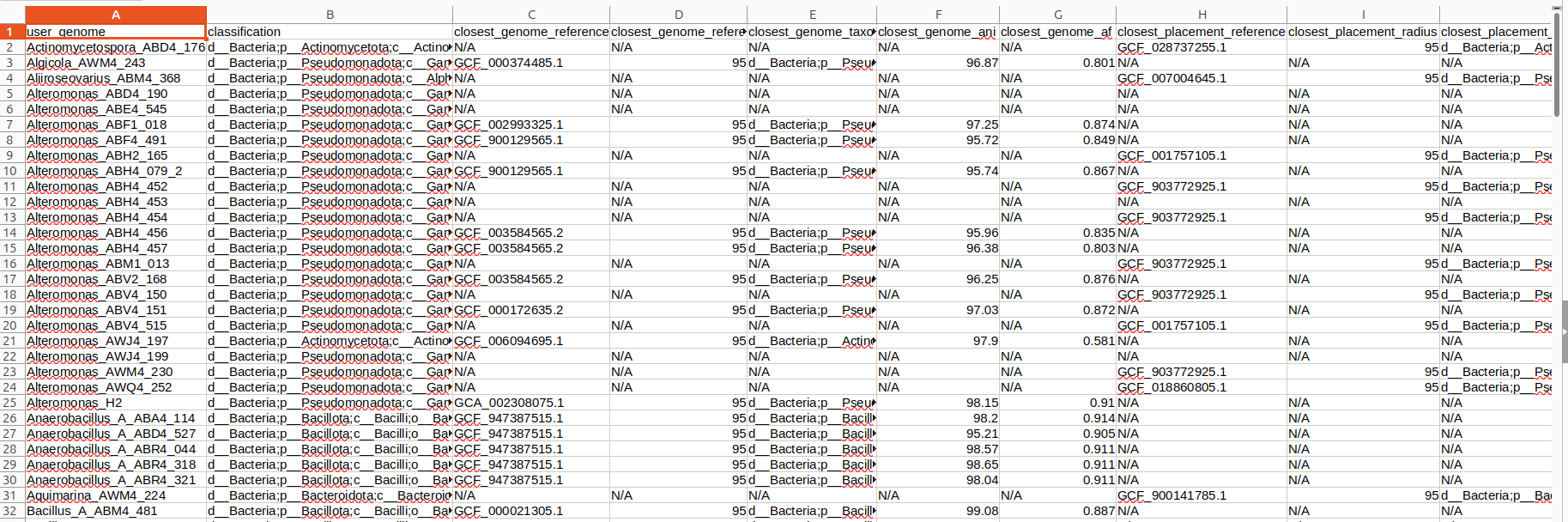
📄 Key output file
gtdbtk.bac120.summary.tsv
This table provides:
• GTDB taxonomy (domain → species)
• Closest reference genomes
• Novelty indicators
• This file becomes the starting point for ANI, AAI, and dDDH analyses.\
• This file is also needed for running Denovo work flow in next step
Below is an example screenshort of gtdbtk.bac120.summary.tsv
4️⃣ Full de_novo_wf (Automatic Tree Building)
For phylogenomic tree construction across many genomes:
gtdbtk de_novo_wf \
--genome_dir genomes/ \
--out_dir gtdbtk/de_novo \
--extension fa \
--bacteria \
--cpus 32 \
--outgroup_taxon p__Chloroflexota \
--skip_gtdb_refs
Notes
Choose an outgroup not present in your dataset
–skip_gtdb_refs reduces tree size for large studies
📄 Output tree:
gtdbtk.bac120.decorated.tree
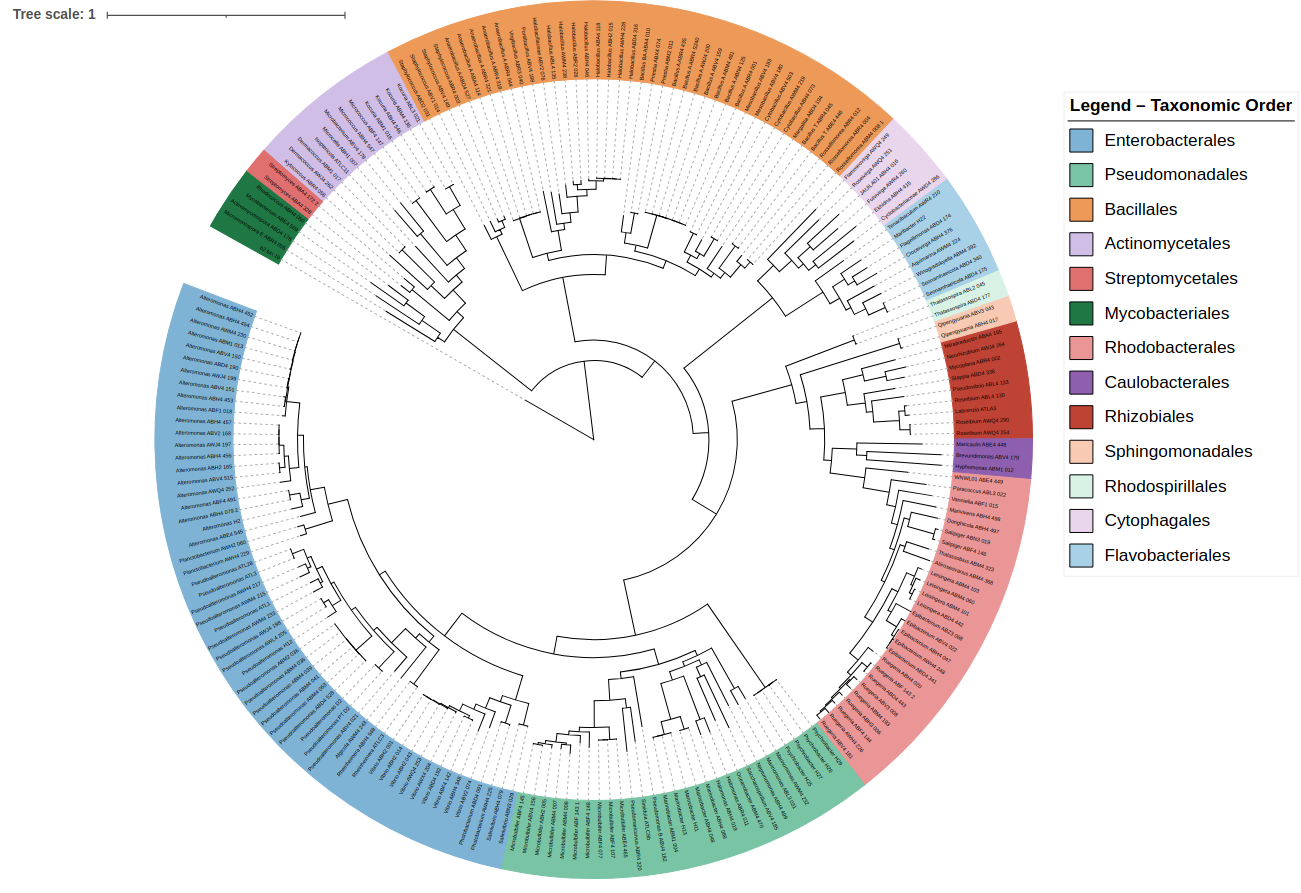
🌳 Tree Visualization with iTOL
Convert GTDB-Tk trees for iTOL:
gtdbtk convert_to_itol \
--input_tree gtdbtk/de_novo/gtdbtk.bac120.decorated.tree \
--output_tree phylogeny_itol/OUTPUT_TREE
Upload to 👉 https://itol.embl.de/
What iTOL allows you to do
✔ Color branches by taxonomy ✔ Add metadata strips (sample type, site, season) ✔ Add heatmaps (genome size, gene counts, FRed, CAZymes) ✔ Collapse or expand clades ✔ Export publication-quality figures (SVG, PDF)
For more deatils on GTDBTk and iTol please see https://jojyjohn28.github.io/blog/gtdbtk-tree-custom/ A video tutorial will be coming soon too. please stay tuned.
🧮 Genome Similarity Metrics (Batch Mode)
Taxonomy alone is not sufficient for species-level conclusions. We therefore apply three complementary genome similarity metrics.
🔢 ANI — Average Nucleotide Identity (FastANI)
✔ Purpose: Species-level resolution
✔ Typical cutoff: ~95–96%
✔ FastANI is fast and suitable for large genome collections.
🔬 AAI — Average Amino Acid Identity (CompareM)
✔ Purpose: Genus- and higher-level resolution
Typical cutoffs:
✔ Species: ~95%
✔ Genus: ~65–75%
✔ AAI is especially useful when ANI is very low or ambiguous.
🧬 dDDH — Digital DNA–DNA Hybridization (GGDC)
Purpose: Classical species validation Species cutoff: 70%
GGDC is currently web-based: 👉 https://ggdc.dsmz.de/
Used primarily for:
✔ Novel species descriptions
✔ MRAs
✔ Formal taxonomic studies
🧠 Why Use ANI + AAI + dDDH Together?
Each metric captures different evolutionary signals:
| Metric | Resolution |
|---|---|
| ANI | Species |
| AAI | Genus and above |
| dDDH | Classical species confirmation |
✔ Agreement across all three = strong taxonomic confidence ✔ Disagreement flags borderline or novel taxa
For more deatails about each analysis and codes can be find at : https://jojyjohn28.github.io/blog/novel-species-a-step-by-step-guide/
🧠 Key Takeaways from Day 3
● Genome-based taxonomy is now the standard ● GTDB-Tk provides reproducible, scalable classification ● Phylogenomic trees add essential evolutionary context ● Species delineation requires multiple similarity metrics ● This workflow scales cleanly to 100+ genomes
🔜 Coming Up: Day 4
Day 4 — Genome Annotation & Functional Potential
We move from who the genome is to what the genome can do — covering gene prediction, functional annotation, and metabolic reconstruction.
All codes can be found at : https://github.com/jojyjohn28/whole-genome-sequencing-analysis