Whole Genome Sequencing — Day 2: Genome Assembly, Quality Assessment, and Topology
🧬 Whole Genome Sequencing — Day 2
Genome Assembly, Quality Assessment, and Topology
In Day 1, we focused on raw read quality control and preprocessing. Today, we move to the next critical step in whole genome analysis:
👉 Genome assembly and assembly evaluation
This step transforms high-quality sequencing reads into contiguous genomic sequences that form the foundation for taxonomy, annotation, and comparative genomics.
🧩 From Reads to Genomes: What Are Reads, Contigs, and Sequences?
Before diving into assembly tools, it’s important to clarify a few core concepts that often cause confusion.
🔹 What are raw reads?
Reads are short (Illumina) or long (Nanopore/PacBio) DNA fragments directly produced by sequencing machines. They represent small, unordered pieces of the genome — like shattered fragments of a book.
• Illumina: typically 100–300 bp
• Nanopore/PacBio: thousands to millions of bp
*On their own, reads do not represent genes or genomes.
🔹 What is a contig?
A contig (contiguous sequence) is created by assembling overlapping reads into a longer continuous DNA sequence.
• Built computationally using assemblers (SPAdes, MEGAHIT, Flye, etc.)
• Represents a reconstructed region of the genome
• Can range from a few kb to several Mb
👉 In draft assemblies, a genome may consist of many contigs.
🔹 What does “sequence” mean in genome assembly?
In practice, a sequence usually refers to:
• a contig, or
• a scaffold, or
• a complete chromosome (single contig)
So when we say “one sequence represents the chromosome”, we mean: ✔ the entire chromosome is assembled into one contig, often circular in bacteria.
##🧠 Assembling a Broken Book
Think of genome assembly like this:
• 📄 Reads → torn sentences
• 📚 Contigs → reconstructed paragraphs
• 📖 Genome → the full book
*Short-read data often leaves gaps (many contigs), while long reads can reconstruct the whole book in one piece. 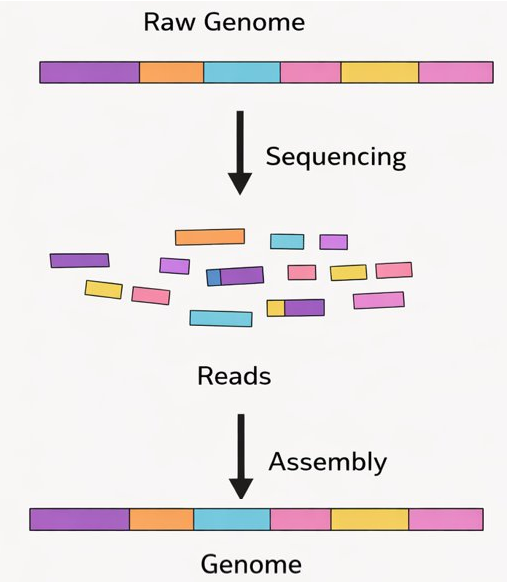
##🔧 Assembly Strategies Overview
Different sequencing technologies require different assembly approaches:
| Data type | Typical tools |
|---|---|
| Illumina short reads | SPAdes, MEGAHIT, Shovill |
| Nanopore / PacBio long reads | Flye |
| Hybrid (short + long) | SPAdes hybrid, Unicycler |
🧱 Short-Read Genome Assembly (Illumina)
1️⃣ SPAdes
SPAdes is widely used for bacterial isolate assemblies and supports multiple modes.
spades.py \
-1 sample_R1.fastq.gz \
-2 sample_R2.fastq.gz \
-o spades_out \
--threads 32 \
--memory 128
Best for: ✔ Isolates ✔ Moderate genome sizes ✔ High accuracy
2️⃣ MEGAHIT
MEGAHIT is optimized for speed and large datasets.
megahit \
-1 sample_R1.fastq.gz \
-2 sample_R2.fastq.gz \
-o megahit_out \
--num-cpu-threads 32
Best for: ✔ Large datasets ✔ Rapid exploratory assemblies ✔ Metagenomes or many isolates
3️⃣ Shovill (Pipeline wrapper)
Shovill is a fast, practical wrapper around SPAdes with automatic preprocessing.
shovill \
--R1 sample_R1.fastq.gz \
--R2 sample_R2.fastq.gz \
--outdir shovill_out \
--cpus 32
For more details about Shovil Assembler ; Please read https://jojyjohn28.github.io/blog/genome-assembly-day/ Why I like Shovill: ✔ Simple ✔ Opinionated defaults ✔ Ideal for batch isolate assembly
🧬 Long-Read Assembly (Nanopore / PacBio)
4️⃣ Flye
Flye is designed specifically for long noisy reads and can resolve complete circular chromosomes.
flye \
--nano-raw sample_nanopore.fastq.gz \
--out-dir flye_out \
--threads 32
Flye often outputs:
✔ Single contig bacterial chromosomes
✔ Assembly graphs indicating circular topology
✔ Coverage estimates
For more details; Please read https://jojyjohn28.github.io/blog/genome-assembly-day/
5️⃣ EPI2ME: Real-Time Analysis for Oxford Nanopore Data
EPI2ME is Oxford Nanopore Technologies’ cloud-based (and increasingly local) analysis platform designed to process Nanopore sequencing data in real time.
Why EPI2ME is useful
•Runs during sequencing (live basecalling + analysis)
•Minimal setup — ideal for quick checks and teaching
Provides:
•Read quality summaries
•Taxonomic classification
•Assembly workflows (selected pipelines)
•AMR and functional screens (workflow-dependent)
Limitations
•Less customizable than command-line workflows
•Not ideal for large comparative projects
•Cloud dependency (unless using EPI2ME Labs locally)
Best practice: Treat EPI2ME as a first look, not a replacement for reproducible, command-line assembly pipelines (Flye, SPAdes, etc.).
🧬 Hybrid Assembly: Combining Short and Long Reads
Modern bacterial genome assembly often benefits from hybrid assembly, which integrates:
•Short reads (Illumina) → high base accuracy
•Long reads (ONT / PacBio) → long-range continuity
Together, they solve problems that each technology alone cannot.
Why hybrid assembly matters | Challenge | Short reads | Long reads | Hybrid | | ——————— | ———– | ———- | —— | | Repeats | ❌ | ✅ | ✅ | | Structural resolution | ❌ | ✅ | ✅ | | Base accuracy | ✅ | ❌ | ✅ | | Circular genomes | ❌ | ✅ | ✅ |
🔧 Common Hybrid Assembly Strategies
1️⃣ Short-read–first (polish later)
•Assemble long reads with Flye
•Polish using Illumina reads (e.g., Pilon)
•Best when long-read coverage is high
2️⃣ True hybrid assemblers
• SPAdes (–hybrid)
• Unicycler (highly recommended for bacteria)_ my favorite
Example (Unicycler):
unicycler \
-1 reads_R1.fastq.gz \
-2 reads_R2.fastq.gz \
-l nanopore.fastq.gz \
-o unicycler_out \
-t 32
🧠 Choosing the Right Strategy | Data available | Recommended approach | | ———————— | ————————– | | Illumina only | SPAdes / Shovill | | ONT only (high coverage) | Flye | | Illumina + ONT | Unicycler or SPAdes hybrid | | Training / rapid checks | EPI2ME |
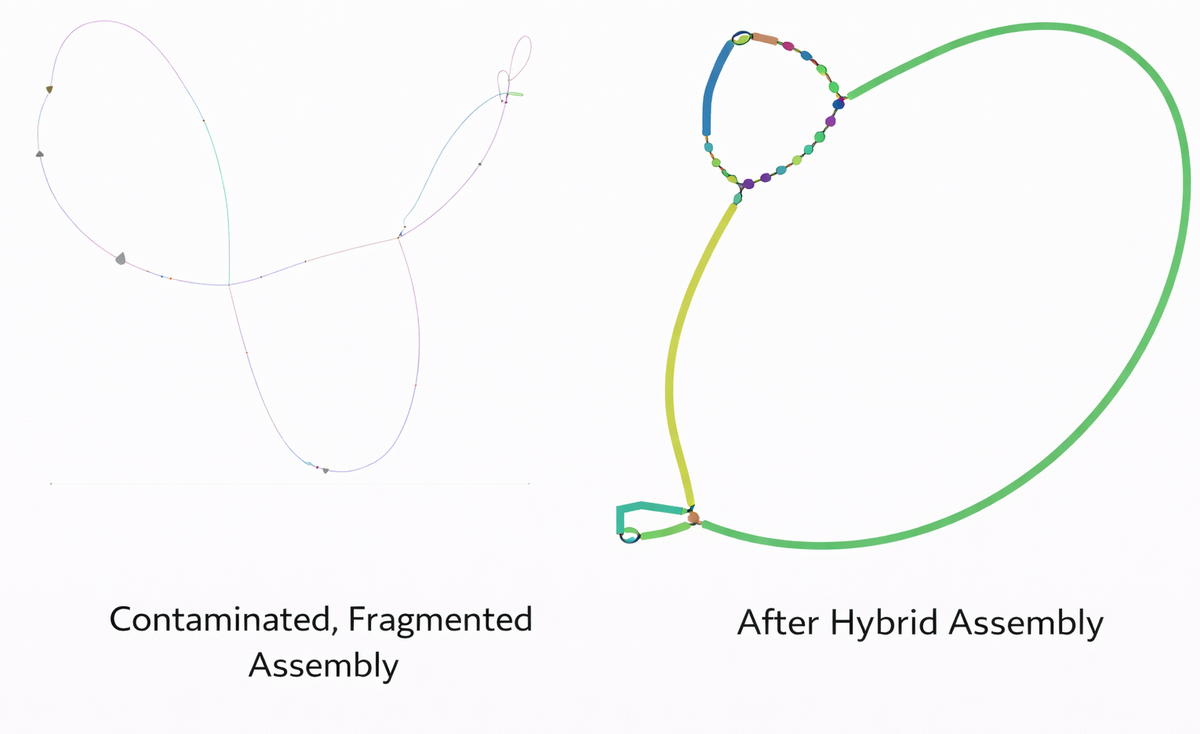 In this image Left panel: fragmented / problematic assembly and Right panel: improved assembly after hybrid approach. The assembly graph is visulized on Bandage.
In this image Left panel: fragmented / problematic assembly and Right panel: improved assembly after hybrid approach. The assembly graph is visulized on Bandage.
📊 Assembly Evaluation with QUAST
Once assemblies are generated, QUAST is used to evaluate their quality.
quast.py \
spades_out/contigs.fasta \
megahit_out/final.contigs.fa \
shovill_out/contigs.fa \
flye_out/assembly.fasta \
-o quast_results
QUAST reports include:
• Genome size
• Number of contigs
• N50 / L50
• GC content
• Presence of Ns
If you have many genomes and plan to submit them to NCBI or a Microbial Resource Announcement, you can extract QUAST results as a summary table using Python 🐍 Example Python script,quast_to_table.py, can be find at day 2 of https://github.com/jojyjohn28/whole-genome-sequencing-analysis
📈 GC Content Visualization (R)
GC content is a quick diagnostic for contamination or misassemblies. Eaxmple R script GC_vis.R also can be find at day 2 of https://github.com/jojyjohn28/whole-genome-sequencing-analysis
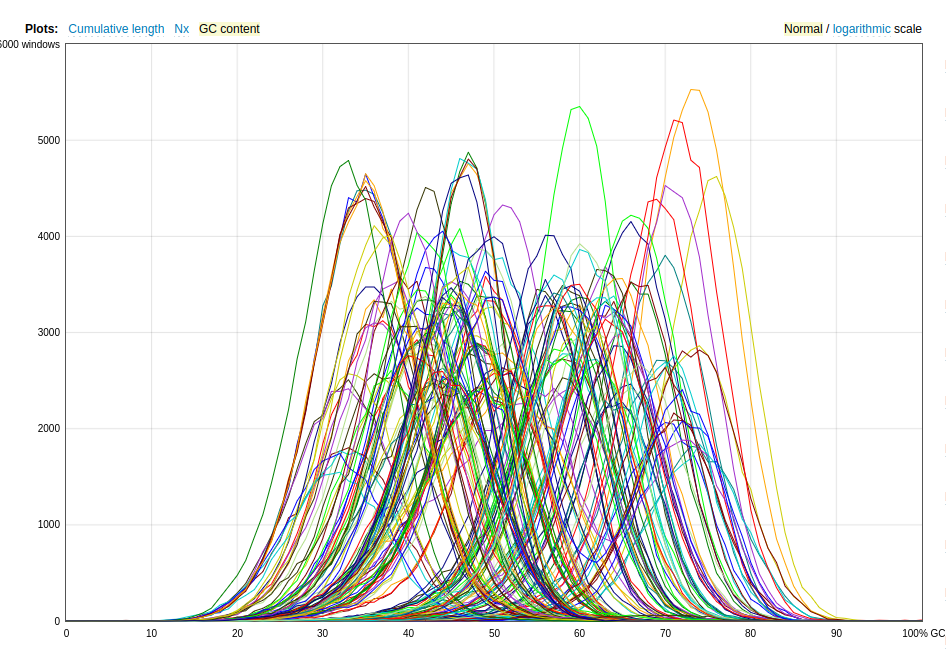
🔁 Genome Topology: Linear vs Circular
Determining topology is essential for:
• NCBI submission
• Genome announcements
• Biological interpretation
General rules:
• Short-read assemblies → treat as linear/draft
• Long-read Flye assemblies → may be circular
To validate circularity:
•Inspect Flye logs
• Check for overlap at contig ends
• Confirm no duplicated terminal regions More details and step by step instruction are published at : https://jojyjohn28.github.io/blog/genome-topology-and-genome-report/
🧠 Assembly Graph Inspection with Bandage
Bandage allows visual inspection of assembly graphs. Assembly graph visulization using bandage can be find at : https://jojyjohn28.github.io/blog/genome-visualization/
🧠 Key Takeaways from Day 2
● No single assembler fits all data ● Always evaluate assemblies with QUAST ● GC content is a powerful sanity check ● Long reads enable topology resolution ● Bandage helps see what statistics cannot
All batch scripts are available in the project repository. https://github.com/jojyjohn28/whole-genome-sequencing-analysis/day2
In Day 3, we will move from assemblies to taxonomy, phylogeny, and species delineation using GTDB-Tk, ANI, AAI, and dDDH.