From QIIME 2 to R — Visualizing Your 16S Amplicon Results
From QIIME 2 to R — visualizing your 16S amplicon results
🧬 𝐷𝑎𝑦 73 𝑜𝑓 𝐷𝑎𝑖𝑙𝑦 𝐵𝑖𝑜𝑖𝑛𝑓𝑜𝑟𝑚𝑎𝑡𝑖𝑐𝑠 𝑓𝑟𝑜𝑚 𝐽𝑜𝑗𝑦’𝑠
You ran your 16S samples through QIIME 2. You have feature tables, taxonomy assignments, a phylogenetic tree, and alpha and beta diversity results. You opened a few .qzv files in the browser, which is great for exploration — but when it comes to making figures for a paper, or for priliminary analysis you want full control in R.
This post is the bridge between your QIIME 2 outputs and your first set of publication-quality figures. We will export every artifact you need, convert it into formats R can read, and then build the figures step by step.
Unlike my earlier 16S series that relied on microeco for downstream visualization, this workflow uses simple, direct R plotting from exported QIIME 2 outputs, which makes it easier to customize figures for papers, reports, blog posts, and LinkedIn.
What we are building: reads retained per sample, ASV counts, alpha diversity (Shannon, observed features, Faith’s PD), beta diversity (Bray-Curtis PCoA + PERMANOVA), and stacked taxonomy barplots at phylum, family, and genus level — all combined into a single multi-panel figure.
What you need before starting
This guide assumes you have already completed the QIIME 2 analysis (import → trim → DADA2 → taxonomy → filtering → core metrics). Your working directory should contain files like these:
table-no-contam_240_200.qza
taxonomy_240_200.qza
denoising-stats_240_200.qza
rooted-tree_240_200.qza
core-metrics-results_50000/
my_metadata.txt
File names will vary by project — adapt them throughout.
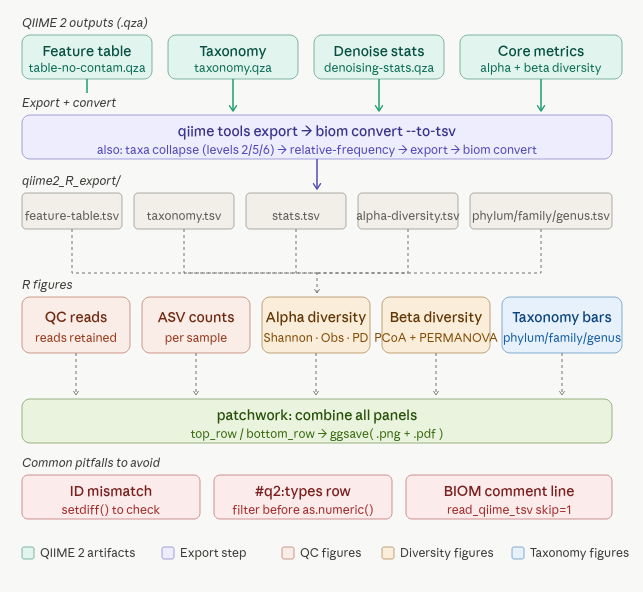
Part 1: Exporting from QIIME 2
QIIME 2 stores everything in .qza and .qzv files, which are essentially zip archives with provenance tracking. Before R can read them, you need to export the underlying data files.
Step 1: Create a clean export directory
Keep all exported files together in one folder. This makes paths in R much simpler.
mkdir -p qiime2_R_export
Step 2: Export the feature table
qiime tools export \
--input-path table-no-contam_240_200.qza \
--output-path qiime2_R_export/table
This creates qiime2_R_export/table/feature-table.biom. BIOM is a compressed binary format — R cannot read it directly, so we convert it to TSV:
biom convert \
-i qiime2_R_export/table/feature-table.biom \
-o qiime2_R_export/table/feature-table.tsv \
--to-tsv
Step 3: Export taxonomy
qiime tools export \
--input-path taxonomy_240_200.qza \
--output-path qiime2_R_export/taxonomy
This produces taxonomy.tsv — a two-column file with feature IDs and their SILVA taxonomy strings.
Step 4: Export denoising statistics
qiime tools export \
--input-path denoising-stats_240_200.qza \
--output-path qiime2_R_export/denoising_stats
This gives you stats.tsv — the per-sample read counts at every DADA2 step (input → filtered → denoised → merged → non-chimeric). This is important for quality checking.
Step 5: Export alpha diversity vectors
qiime tools export \
--input-path core-metrics-results_50000/shannon_vector.qza \
--output-path qiime2_R_export/shannon
qiime tools export \
--input-path core-metrics-results_50000/observed_features_vector.qza \
--output-path qiime2_R_export/observed_features
qiime tools export \
--input-path core-metrics-results_50000/faith_pd_vector.qza \
--output-path qiime2_R_export/faith_pd
Each of these exports a simple two-column TSV: sample ID and diversity value.
Step 6: Export beta diversity outputs
qiime tools export \
--input-path core-metrics-results_50000/bray_curtis_distance_matrix.qza \
--output-path qiime2_R_export/bray_distance
qiime tools export \
--input-path core-metrics-results_50000/bray_curtis_pcoa_results.qza \
--output-path qiime2_R_export/bray_pcoa
Step 7: Collapse taxonomy and convert to relative abundance
For stacked barplots, you want the feature table aggregated at phylum, family, and genus level. QIIME 2 does this cleanly with taxa collapse.
SILVA taxonomy levels:
- Level 2 = Phylum
- Level 5 = Family
- Level 6 = Genus
# Collapse
qiime taxa collapse \
--i-table table-no-contam_240_200.qza \
--i-taxonomy taxonomy_240_200.qza \
--p-level 2 \
--o-collapsed-table phylum.qza
qiime taxa collapse \
--i-table table-no-contam_240_200.qza \
--i-taxonomy taxonomy_240_200.qza \
--p-level 5 \
--o-collapsed-table family.qza
qiime taxa collapse \
--i-table table-no-contam_240_200.qza \
--i-taxonomy taxonomy_240_200.qza \
--p-level 6 \
--o-collapsed-table genus.qza
# Convert to relative abundance
qiime feature-table relative-frequency --i-table phylum.qza --o-relative-frequency-table phylum_rel.qza
qiime feature-table relative-frequency --i-table family.qza --o-relative-frequency-table family_rel.qza
qiime feature-table relative-frequency --i-table genus.qza --o-relative-frequency-table genus_rel.qza
# Export
qiime tools export --input-path phylum_rel.qza --output-path qiime2_R_export/phylum
qiime tools export --input-path family_rel.qza --output-path qiime2_R_export/family
qiime tools export --input-path genus_rel.qza --output-path qiime2_R_export/genus
# Convert BIOM to TSV for each
cd qiime2_R_export
biom convert -i phylum/feature-table.biom -o phylum/feature-table.tsv --to-tsv
biom convert -i family/feature-table.biom -o family/feature-table.tsv --to-tsv
biom convert -i genus/feature-table.biom -o genus/feature-table.tsv --to-tsv
Step 8: Copy your metadata
cp my_metadata.txt qiime2_R_export/
What your export folder should look like
qiime2_R_export/
├── table/
│ ├── feature-table.biom
│ └── feature-table.tsv
├── taxonomy/
│ └── taxonomy.tsv
├── denoising_stats/
│ └── stats.tsv
├── shannon/
│ └── alpha-diversity.tsv
├── observed_features/
│ └── alpha-diversity.tsv
├── faith_pd/
│ └── alpha-diversity.tsv
├── bray_distance/
│ └── distance-matrix.tsv
├── bray_pcoa/
│ └── ordination.txt
├── phylum/
│ └── feature-table.tsv
├── family/
│ └── feature-table.tsv
├── genus/
│ └── feature-table.tsv
└── my_metadata.txt
Part 2: Visualization in R
Set your working directory to the export folder first:
setwd("path/to/qiime2_R_export")
Libraries
library(tidyverse) # data manipulation + ggplot2
library(vegan) # PERMANOVA (adonis2)
library(biomformat) # read BIOM files if needed
library(viridis) # colour palettes
library(patchwork) # combine multiple plots
library(cowplot) # draw_label for text panels
A: Read metadata
Your metadata file is the anchor that connects every other file. Every sample ID in every TSV must match the IDs here exactly.
meta <- read.delim("my_metadata.txt", sep = "\t", check.names = FALSE)
colnames(meta)[1] <- "SampleID"
# Set a sensible factor order for treatments
meta$Treatment <- factor(meta$Treatment, levels = c("NC", "NPK", "nOB9"))
B: Denoising statistics — how many reads did DADA2 keep?
denoise <- read.delim("denoising_stats/stats.tsv",
sep = "\t", check.names = FALSE, stringsAsFactors = FALSE)
# Clean column names (QIIME2 uses hyphens and spaces)
colnames(denoise) <- gsub("[- ]", "_", colnames(denoise))
colnames(denoise)[1] <- "SampleID"
# Remove the QIIME2 type annotation row
denoise <- denoise[denoise$SampleID != "#q2:types", ]
# Convert counts to numeric
num_cols <- setdiff(colnames(denoise), "SampleID")
denoise[num_cols] <- lapply(denoise[num_cols], as.numeric)
Why the #q2:types row? QIIME 2 metadata files include a second header row that describes data types (e.g. numeric, categorical). It looks like data but it is metadata about the metadata. Always remove it before any numeric conversion.
reads_plot <- ggplot(denoise, aes(x = SampleID, y = non_chimeric, fill = SampleID)) +
geom_col() +
theme_bw() +
labs(title = "Reads retained after DADA2", x = NULL, y = "Non-chimeric reads") +
theme(axis.text.x = element_text(angle = 45, hjust = 1), legend.position = "none")
C: ASV counts per sample
library(biomformat)
table_biom <- read_biom("table/feature-table.biom")
asv_mat <- as.data.frame(as.matrix(biom_data(table_biom)))
asv_per_sample <- colSums(asv_mat)
asv_df <- data.frame(SampleID = names(asv_per_sample), ASV_count = asv_per_sample)
asv_plot <- ggplot(asv_df, aes(x = SampleID, y = ASV_count, fill = SampleID)) +
geom_col() +
theme_bw() +
labs(title = "Total ASV abundance per sample", x = NULL, y = "Total ASV abundance") +
theme(axis.text.x = element_text(angle = 45, hjust = 1), legend.position = "none")
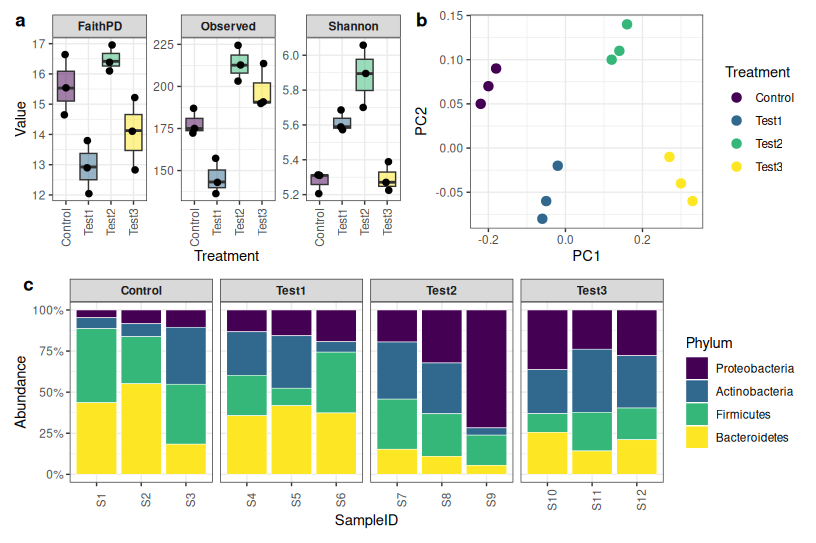
D: Alpha diversity — Shannon, observed features, Faith’s PD
shannon <- read.delim("shannon/alpha-diversity.tsv", sep = "\t")
obs <- read.delim("observed_features/alpha-diversity.tsv", sep = "\t")
faith <- read.delim("faith_pd/alpha-diversity.tsv", sep = "\t")
colnames(shannon) <- c("SampleID", "Shannon")
colnames(obs) <- c("SampleID", "Observed")
colnames(faith) <- c("SampleID", "FaithPD")
alpha_df <- meta %>%
left_join(shannon, by = "SampleID") %>%
left_join(obs, by = "SampleID") %>%
left_join(faith, by = "SampleID")
Plot all three metrics on one faceted panel:
alpha_long <- alpha_df %>%
pivot_longer(cols = c(Shannon, Observed, FaithPD),
names_to = "Metric", values_to = "Value")
alpha_plot <- ggplot(alpha_long, aes(x = Treatment, y = Value, fill = Treatment)) +
geom_boxplot(alpha = 0.5, outlier.shape = NA) +
geom_jitter(width = 0.1, size = 2) +
facet_wrap(~ Metric, scales = "free_y") +
scale_fill_viridis_d(option = "plasma") +
theme_bw() +
labs(title = "Alpha diversity", x = NULL, y = NULL) +
theme(strip.text = element_text(face = "bold"), legend.position = "none")
Optional: statistical testing
kruskal.test(Shannon ~ Treatment, data = alpha_df)
kruskal.test(Observed ~ Treatment, data = alpha_df)
kruskal.test(FaithPD ~ Treatment, data = alpha_df)
# Pairwise comparisons
pairwise.wilcox.test(alpha_df$Shannon, alpha_df$Treatment, p.adjust.method = "BH")
What these metrics mean:
- Shannon index — accounts for both richness (how many taxa) and evenness (how evenly distributed). Higher = more diverse.
- Observed features — simply the count of unique ASVs detected. Sensitive to sampling depth.
- Faith’s PD — phylogenetic diversity. Sums the branch lengths of the phylogenetic tree covered by the detected ASVs. Higher = more evolutionary breadth.
E: Beta diversity — Bray-Curtis PCoA + PERMANOVA
Reading the PCoA ordination file requires a small trick: QIIME 2 writes a header with metadata about eigenvalues above the coordinate data. We skip 4 lines to get to the actual coordinates.
pcoa <- read.delim("bray_pcoa/ordination.txt",
sep = "\t", skip = 4, check.names = FALSE,
row.names = 1)
# Keep only sample rows (not the "Site" or "Species" annotation rows)
coords <- pcoa[!(rownames(pcoa) %in% c("Species", "Site")), 1:2, drop = FALSE]
coords$SampleID <- rownames(coords)
colnames(coords) <- c("PC1", "PC2", "SampleID")
coords$PC1 <- as.numeric(coords$PC1)
coords$PC2 <- as.numeric(coords$PC2)
ord_df <- left_join(coords, meta, by = "SampleID")
PERMANOVA tests whether treatment groups have significantly different community composition:
dist_mat <- read.delim("bray_distance/distance-matrix.tsv",
sep = "\t", row.names = 1, check.names = FALSE)
dist_obj <- as.dist(dist_mat)
adon <- adonis2(dist_obj ~ Treatment, data = meta, permutations = 999)
pval <- adon$`Pr(>F)`[1]
r2 <- adon$R2[1]
beta_plot <- ggplot(ord_df, aes(PC1, PC2, color = Treatment)) +
geom_point(size = 3) +
stat_ellipse() +
theme_bw() +
labs(title = paste0("Bray-Curtis PCoA\nPERMANOVA p=", signif(pval, 3),
", R²=", round(r2, 3)),
x = "PC1", y = "PC2")
What PERMANOVA R² means: the proportion of community variation explained by the grouping variable. R² = 0.35 means 35% of the variation between samples is associated with treatment.
Pairwise PERMANOVA (which specific treatments differ from each other?):
pairwise_adonis <- function(dist, factors) {
comb <- combn(unique(factors), 2)
results <- data.frame()
for (i in 1:ncol(comb)) {
group <- comb[, i]
idx <- factors %in% group
sub_dist <- as.dist(as.matrix(dist)[idx, idx])
sub_meta <- data.frame(Treatment = factors[idx])
ad <- adonis2(sub_dist ~ Treatment, data = sub_meta)
results <- rbind(results, data.frame(Group1=group[1], Group2=group[2],
R2=ad$R2[1], p=ad$`Pr(>F)`[1]))
}
results$p_adj <- p.adjust(results$p, method = "BH")
results
}
pairwise_adonis(dist_obj, meta$Treatment)
F: Taxonomy helper functions
Two small functions make all the taxonomy plots reusable.
Helper 1: Read a QIIME 2 TSV
read_qiime_tsv <- function(path) {
x <- read.delim(path, sep = "\t", header = TRUE,
comment.char = "", check.names = FALSE,
stringsAsFactors = FALSE, skip = 1)
colnames(x)[1] <- "Taxon"
x
}
The skip = 1 is important — BIOM-converted TSVs start with a comment line (# Constructed from biom file) that is not data.
Helper 2: Clean SILVA taxonomy strings
SILVA taxonomy strings look like: d__Bacteria;p__Proteobacteria;c__Gammaproteobacteria;.... The function below pulls out the level you want and strips the prefix.
extract_tax_label <- function(taxon_string, level = c("phylum","family","genus")) {
level <- match.arg(level)
sapply(taxon_string, function(x) {
parts <- trimws(unlist(strsplit(x, ";")))
val <- switch(level,
phylum = parts[2],
family = parts[5],
genus = parts[6])
if (length(val) == 0 || is.na(val) || val == "") return("Unassigned")
val <- sub("^[a-z]__", "", val)
if (val %in% c("", "uncultured", "metagenome", "Ambiguous_taxa")) return("Unassigned")
val
})
}
Helper 3: Build a long-format taxonomy table
make_long_taxa <- function(path, meta, level = c("phylum","family","genus")) {
level <- match.arg(level)
tab <- read_qiime_tsv(path)
tab %>%
pivot_longer(-Taxon, names_to = "SampleID", values_to = "Abundance") %>%
mutate(Abundance = as.numeric(Abundance),
Label = extract_tax_label(Taxon, level = level)) %>%
group_by(SampleID, Label) %>%
summarise(Abundance = sum(Abundance, na.rm = TRUE), .groups = "drop") %>%
left_join(meta, by = "SampleID")
}
G: Stacked taxonomy barplots
plot_taxa_stacked <- function(long_df, title_txt, top_n = 15) {
# Identify top N taxa by total abundance
top_taxa <- long_df %>%
group_by(Label) %>%
summarise(Total = sum(Abundance, na.rm = TRUE), .groups = "drop") %>%
arrange(desc(Total)) %>%
slice_head(n = top_n) %>%
pull(Label)
# Merge everything else into "Other"
plot_df <- long_df %>%
mutate(Label = ifelse(Label %in% top_taxa, Label, "Other")) %>%
group_by(SampleID, Treatment, Label) %>%
summarise(Abundance = sum(Abundance, na.rm = TRUE), .groups = "drop")
# Order fill by overall abundance
levs <- plot_df %>%
group_by(Label) %>%
summarise(Total = sum(Abundance), .groups = "drop") %>%
arrange(desc(Total)) %>% pull(Label)
plot_df$Label <- factor(plot_df$Label, levels = rev(unique(levs)))
plot_df$SampleID <- factor(plot_df$SampleID, levels = levels(meta$SampleID))
plot_df$Treatment <- factor(plot_df$Treatment, levels = levels(meta$Treatment))
ggplot(plot_df, aes(x = SampleID, y = Abundance, fill = Label)) +
geom_col(width = 0.9, colour = "white", linewidth = 0.1) +
scale_fill_viridis_d(option = "turbo") +
scale_y_continuous(labels = function(x) paste0(round(x * 100), "%")) +
facet_grid(. ~ Treatment, scales = "free_x", space = "free_x") +
labs(title = title_txt, x = "Sample", y = "Relative abundance", fill = NULL) +
theme_bw() +
theme(axis.text.x = element_text(angle = 90, vjust = 0.5, hjust = 1),
panel.grid = element_blank(),
strip.background = element_rect(fill = "white"),
strip.text = element_text(face = "bold"))
}
# Build the three tables
phylum_long <- make_long_taxa("phylum/feature-table.tsv", meta, "phylum")
family_long <- make_long_taxa("family/feature-table.tsv", meta, "family")
genus_long <- make_long_taxa("genus/feature-table.tsv", meta, "genus")
# Build the three plots
phylum_plot <- plot_taxa_stacked(phylum_long, "Phylum-level relative abundance", top_n = 12)
family_plot <- plot_taxa_stacked(family_long, "Family-level relative abundance", top_n = 20)
genus_plot <- plot_taxa_stacked(genus_long, "Genus-level relative abundance", top_n = 20)
H: Text summary panel
This creates a plain-text summary card that works well in the top-left corner of a multi-panel figure.
total_input <- sum(denoise$input, na.rm = TRUE)
total_nonchim <- sum(denoise$non_chimeric, na.rm = TRUE)
avg_nonchim <- mean(denoise$non_chimeric, na.rm = TRUE)
summary_text <- ggdraw() +
draw_label(
paste0(
"16S Amplicon Summary\n\n",
"Total input reads: ", format(total_input, big.mark = ","), "\n",
"Retained reads: ", format(total_nonchim, big.mark = ","), "\n",
"Average per sample: ", round(avg_nonchim, 0), "\n",
"Samples: ", nrow(meta), "\n",
"ASVs detected: ", nrow(asv_mat)
),
x = 0.05, hjust = 0, vjust = 1, size = 12
)
I: Combine everything into one figure
top_row <- summary_text | reads_plot | asv_plot | alpha_plot | beta_plot
bottom_row <- phylum_plot | family_plot | genus_plot
final_plot <- top_row / bottom_row + plot_layout(heights = c(1, 1.1))
ggsave("reference_style_figure.png", final_plot, width = 22, height = 12, dpi = 300)
ggsave("reference_style_figure.pdf", final_plot, width = 22, height = 12)
Part 3: Bonus — taxon-focused plots
Sometimes you want to zoom into a specific group. In the Bacillus inoculant study, the question was whether different treatments altered the abundance of rhizobia — the nitrogen-fixing bacteria.
Rhizobia genera plot
rhizobia_genera <- c(
"Rhizobium", "Bradyrhizobium", "Mesorhizobium", "Ensifer",
"Sinorhizobium", "Azorhizobium", "Allorhizobium", "Neorhizobium",
"Pararhizobium", "Shinella", "Devosia", "Microvirga"
)
# Keep only rhizobia rows
rhizobia_only <- genus_long %>%
filter(Label %in% rhizobia_genera) %>%
group_by(SampleID, Treatment, Label) %>%
summarise(Abundance = sum(Abundance, na.rm = TRUE), .groups = "drop")
rhizobia_only$SampleID <- factor(rhizobia_only$SampleID, levels = levels(meta$SampleID))
rhizobia_only$Treatment <- factor(rhizobia_only$Treatment, levels = levels(meta$Treatment))
# Custom colours so "Other" is always grey
taxa_no_other <- unique(rhizobia_only$Label)
cols <- setNames(viridis::viridis(length(taxa_no_other), option = "turbo"), taxa_no_other)
rhizobia_plot <- ggplot(rhizobia_only,
aes(x = SampleID, y = Abundance, fill = Label)) +
geom_col(width = 0.9, colour = "white", linewidth = 0.1) +
scale_fill_manual(values = cols) +
scale_y_continuous(labels = scales::percent_format(accuracy = 0.01)) +
facet_grid(. ~ Treatment, scales = "free_x", space = "free_x") +
labs(title = "Rhizobia-related genera (zoomed)", x = "Sample",
y = "Relative abundance", fill = NULL) +
theme_bw() +
theme(axis.text.x = element_text(angle = 90, vjust = 0.5, hjust = 1),
panel.grid = element_blank(),
strip.text = element_text(face = "bold"))
Total rhizobia abundance per treatment (boxplot)
rhizobia_total <- genus_long %>%
mutate(is_rhizobia = Label %in% rhizobia_genera) %>%
group_by(SampleID, Treatment) %>%
summarise(Rhizobia = sum(Abundance[is_rhizobia]), .groups = "drop")
ggplot(rhizobia_total, aes(Treatment, Rhizobia, fill = Treatment)) +
geom_boxplot(alpha = 0.5) +
geom_jitter(width = 0.1, size = 2) +
scale_y_continuous(labels = scales::percent) +
theme_bw() +
labs(title = "Total rhizobia abundance by treatment")
Common beginner mistakes
Metadata IDs do not match sample IDs in the table
This is the most common silent failure. Your left_join(coords, meta, by = "SampleID") will produce rows full of NA if IDs differ even by a trailing space or capital letter. Always run:
setdiff(colnames(asv_mat), meta$SampleID) # in table but not in metadata
setdiff(meta$SampleID, colnames(asv_mat)) # in metadata but not in table
Both should return character(0). If not, fix the metadata file or use trimws() on your IDs.
The #q2:types row breaks numeric conversion
QIIME 2 metadata files include a second header row describing types. It looks like a data row but contains strings like "numeric" or "categorical". Always filter it out before calling as.numeric().
denoise <- denoise[denoise$SampleID != "#q2:types", ]
BIOM-to-TSV conversion produces a comment line
The first line of a BIOM-converted TSV is # Constructed from biom file. The read_qiime_tsv() helper above uses skip = 1 to ignore it. If you use read.delim() directly without skipping, R will read this comment as the header — and all your column names will be wrong.
PCoA ordination file has non-sample rows
The QIIME 2 ordination file has rows called "Species" and "Site" below the coordinate data. Always filter them out:
coords <- pcoa[!(rownames(pcoa) %in% c("Species", "Site")), ]
Stacked barplots with raw counts instead of relative abundance
Make sure you used phylum_rel.qza (after qiime feature-table relative-frequency) and not the raw phylum.qza. Raw counts make barplots uninterpretable when samples have different total read depths.
Summary: what does each output file contain?
| File | What is in it | R use |
|---|---|---|
table/feature-table.tsv | ASVs × samples raw counts | Count checks, richness |
taxonomy/taxonomy.tsv | Feature ID → SILVA taxonomy | Taxonomy parsing |
denoising_stats/stats.tsv | Per-sample DADA2 read retention | QC plot |
shannon/alpha-diversity.tsv | Shannon index per sample | Alpha plot |
observed_features/alpha-diversity.tsv | ASV count per sample (rarefied) | Alpha plot |
faith_pd/alpha-diversity.tsv | Phylogenetic diversity per sample | Alpha plot |
bray_distance/distance-matrix.tsv | Pairwise Bray-Curtis distances | PERMANOVA |
bray_pcoa/ordination.txt | PCoA coordinates | Beta diversity plot |
phylum/feature-table.tsv | Relative abundance collapsed to phylum | Stacked barplot |
family/feature-table.tsv | Relative abundance collapsed to family | Stacked barplot |
genus/feature-table.tsv | Relative abundance collapsed to genus | Stacked barplot |
Statistics
You can also do simple statistics on the diversity analysis like below.
############################################################
# Simple stats for alpha and beta diversity
############################################################
library(tidyverse)
library(vegan)
# ==========================
# 1. Load data
# ==========================
# Replace with your paths
meta <- read.delim("my_metadata.txt", sep = "\t")
shannon <- read.delim("shannon/alpha-diversity.tsv", sep = "\t")
observed <- read.delim("observed_features/alpha-diversity.tsv", sep = "\t")
faith <- read.delim("faith_pd/alpha-diversity.tsv", sep = "\t")
colnames(shannon) <- c("SampleID", "Shannon")
colnames(observed) <- c("SampleID", "Observed")
colnames(faith) <- c("SampleID", "FaithPD")
# Merge
alpha_df <- meta %>%
left_join(shannon, by = "SampleID") %>%
left_join(observed, by = "SampleID") %>%
left_join(faith, by = "SampleID")
alpha_df$Treatment <- factor(alpha_df$Treatment)
# ==========================
# 2. Alpha diversity stats
# ==========================
# ---- Kruskal-Wallis (global)
kw_shannon <- kruskal.test(Shannon ~ Treatment, data = alpha_df)
kw_observed <- kruskal.test(Observed ~ Treatment, data = alpha_df)
kw_faith <- kruskal.test(FaithPD ~ Treatment, data = alpha_df)
kw_shannon
kw_observed
kw_faith
# ---- Pairwise Wilcoxon
pw_shannon <- pairwise.wilcox.test(alpha_df$Shannon, alpha_df$Treatment, p.adjust.method = "BH")
pw_observed <- pairwise.wilcox.test(alpha_df$Observed, alpha_df$Treatment, p.adjust.method = "BH")
pw_faith <- pairwise.wilcox.test(alpha_df$FaithPD, alpha_df$Treatment, p.adjust.method = "BH")
pw_shannon
pw_observed
pw_faith
# ==========================
# 3. Beta diversity stats
# ==========================
# Load Bray-Curtis distance matrix
dist_mat <- read.delim("bray_distance/distance-matrix.tsv",
sep = "\t", row.names = 1)
dist_obj <- as.dist(dist_mat)
# ---- PERMANOVA
adon <- adonis2(dist_obj ~ Treatment, data = meta, permutations = 999)
adon
# Extract values
pval <- adon$`Pr(>F)`[1]
r2 <- adon$R2[1]
cat("PERMANOVA: R2 =", r2, "p =", pval, "\n")
# ==========================
# 4. Pairwise PERMANOVA
# ==========================
pairwise_adonis <- function(dist, factors) {
comb <- combn(unique(factors), 2)
results <- data.frame()
for(i in 1:ncol(comb)){
group <- comb[,i]
idx <- factors %in% group
sub_dist <- as.dist(as.matrix(dist)[idx, idx])
sub_meta <- data.frame(Treatment = factors[idx])
ad <- adonis2(sub_dist ~ Treatment, data = sub_meta)
results <- rbind(results, data.frame(
Group1 = group[1],
Group2 = group[2],
R2 = ad$R2[1],
p = ad$`Pr(>F)`[1]
))
}
results$p_adj <- p.adjust(results$p, method = "BH")
results
}
pairwise_results <- pairwise_adonis(dist_obj, meta$Treatment)
pairwise_results
# ==========================
# 5. Dispersion test (IMPORTANT)
# ==========================
disp <- betadisper(dist_obj, meta$Treatment)
anova_disp <- anova(disp)
anova_disp
# ==========================
# 6. Save summary tables
# ==========================
alpha_summary <- data.frame(
Metric = c("Shannon", "Observed", "FaithPD"),
p_value = c(kw_shannon$p.value, kw_observed$p.value, kw_faith$p.value)
)
write.csv(alpha_summary, "alpha_stats.csv", row.names = FALSE)
write.csv(pairwise_results, "pairwise_permanova.csv", row.names = FALSE)
Repository
For detailed R scripts and other files see this Repository
This workflow covers the Bacillus inoculant soil microbiome project (16S V4 amplicons, 9 samples across NC/NPK/nOB9 treatments, SILVA 138, DADA2 pseudo-pooling, sampling depth 50,000 reads). The same pipeline applies to any QIIME 2 amplicon dataset — adapt file names and taxonomy level labels to your study.
Tags: QIIME2 16S amplicon R ggplot2 alpha-diversity beta-diversity taxonomy DADA2 PERMANOVA microbiome beginner