| Estimated Time: 8-12 hours | Difficulty: Advanced | Prerequisites: Day 5 (Annotation) |
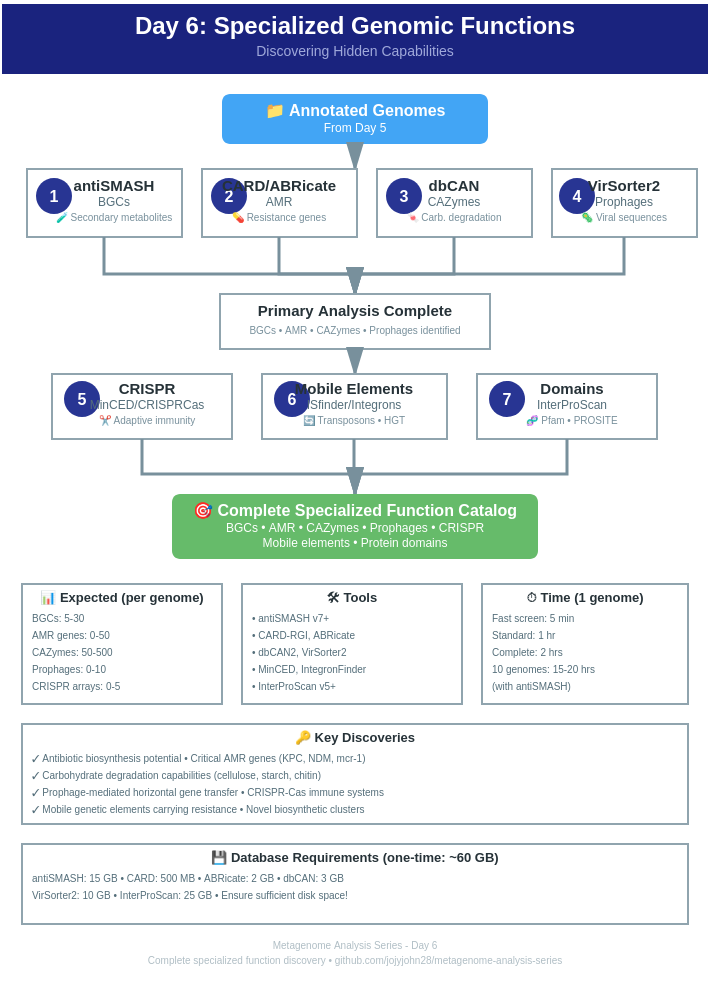
Welcome to Day 6! After basic annotation (Day 5), it’s time to discover hidden genomic treasures - specialized functions that make organisms unique.
Today’s discoveries:
Basic annotation tells you:
Specialized analysis reveals:
antiSMASH (antibiotics & Secondary Metabolite Analysis SHell) is the gold standard for BGC detection.
# Create environment
conda create -n antismash -c bioconda antismash
conda activate antismash
# Download databases (~15 GB)
download-antismash-databases
conda activate antismash
# Single genome
antismash \
--output-dir antismash_output/genome1 \
--genefinding-tool prodigal \
--cpus 8 \
genome1.gbk
# With all features enabled
antismash \
--output-dir antismash_output/genome1 \
--genefinding-tool prodigal \
--knownclusterblast \
--subclusterblast \
--clusterblast \
--smcog-trees \
--cpus 8 \
genome1.gbk
Input formats:
antismash_output/genome1/
├── genome1.gbk # Annotated genome
├── genome1.json # Machine-readable results
├── index.html # Interactive visualization
├── regions.js # BGC regions
├── knownclusterblast/ # Hits to known BGCs
└── svg/ # Cluster visualizations
BGC types detected:
BiG-SCAPE compares BGCs across multiple genomes to identify families.
conda create -n bigscape -c bioconda bigscape
conda activate bigscape
# Collect all BGC GenBank files from antiSMASH
mkdir bigscape_input
cp antismash_output/*/regions/*.gbk bigscape_input/
# Run BiG-SCAPE
bigscape \
-i bigscape_input \
-o bigscape_output \
--pfam_dir ~/pfam_database \
--cores 8 \
--mode auto
# Visualize
firefox bigscape_output/network_files/index.html
BiG-SCAPE output:
RGI (Resistance Gene Identifier) uses CARD database for AMR detection.
conda create -n rgi -c bioconda rgi
conda activate rgi
# Download CARD database
rgi load --card_json ~/card_database/card.json
# Verify
rgi database --version
conda activate rgi
# Predict AMR from proteins
rgi main \
--input_sequence proteins.faa \
--input_type protein \
--output_file rgi_output \
--clean \
--num_threads 8
# From nucleotides (slower but more sensitive)
rgi main \
--input_sequence genome.fa \
--input_type contig \
--output_file rgi_output \
--clean \
--num_threads 8 \
--include_loose
RGI output columns:
ABRicate screens multiple AMR databases quickly.
conda create -n abricate -c bioconda abricate
conda activate abricate
# Update databases
abricate-get_db --db all
conda activate abricate
# Screen against all databases
abricate genome.fa > abricate_results.tab
# Specific database
abricate --db card genome.fa > card_results.tab
abricate --db resfinder genome.fa > resfinder_results.tab
abricate --db vfdb genome.fa > virulence_results.tab
# Batch processing
abricate genomes/*.fa > all_genomes_amr.tab
# Summary
abricate --summary all_genomes_amr.tab > amr_summary.tab
Available databases:
dbCAN identifies enzymes that degrade, modify, or create carbohydrates.
conda create -n dbcan -c bioconda dbcan
conda activate dbcan
# Download databases
dbcan_build --help
conda activate dbcan
# Run dbCAN
run_dbcan \
proteins.faa \
protein \
--out_dir dbcan_output \
--db_dir ~/dbcan_db \
--tools all \
--threads 8
# Output: dbcan_output/overview.txt
CAZyme families:
VirSorter2 identifies viral sequences in genomes and metagenomes.
conda create -n virsorter2 -c bioconda virsorter=2
conda activate virsorter2
# Setup database
virsorter setup -d ~/virsorter2-db -j 4
conda activate virsorter2
# Run VirSorter2
virsorter run \
-i genome.fa \
-w virsorter2_output \
--min-length 5000 \
--min-score 0.5 \
-j 8 \
all
# High-confidence viral sequences
cat virsorter2_output/final-viral-boundary.tsv
VirSorter2 output:
PHASTER identifies prophages with detailed annotation.
Note: PHASTER is primarily a web service.
Web interface: https://phaster.ca/
Alternative: Use VirSorter2 + CheckV
# CheckV for viral genome quality
conda create -n checkv -c bioconda checkv
conda activate checkv
# Download database
checkv download_database ~/checkv-db
# Run CheckV on VirSorter2 results
checkv end_to_end \
virsorter2_output/final-viral-combined.fa \
checkv_output \
-d ~/checkv-db \
-t 8
# Installation
conda create -n crisprcasfinder -c bioconda crisprcasfinder
conda activate crisprcasfinder
# Run
CRISPRCasFinder.pl \
-in genome.fa \
-out crispr_output \
-cas \
-keep
conda create -n minced -c bioconda minced
conda activate minced
# Quick CRISPR detection
minced genome.fa crispr_output.txt
# Parse results
grep "CRISPR" crispr_output.txt
CRISPR system types:
# Use ABRicate with custom IS database
abricate --db isfinder genome.fa > is_results.tab
# Or BLAST against ISfinder database
blastn \
-query genome.fa \
-db ~/isfinder_db/ISfinder \
-outfmt 6 \
-num_threads 8 \
> is_blast.out
conda create -n integron_finder -c bioconda integron_finder
conda activate integron_finder
# Find integrons
integron_finder \
--local-max \
--cpu 8 \
genome.fa
Integrons contain:
InterProScan searches multiple protein signature databases.
# Large download - recommend using web service
# Or install locally:
wget ftp://ftp.ebi.ac.uk/pub/software/unix/iprscan/5/5.XX-XX.X/interproscan-5.XX-XX.X-64-bit.tar.gz
tar -xzf interproscan-5.XX-XX.X-64-bit.tar.gz
# Run InterProScan
interproscan.sh \
-i proteins.faa \
-f TSV,GFF3 \
-o interproscan_output \
--cpu 8 \
--goterms \
--pathways
Databases searched:
Use comprehensive_analysis.sh from Day 6 →
Use compare_specialized_functions.py from Day 6 →
# Check input format
head genome.gbk
# Try with relaxed detection
antismash --minimal ...
# Ensure genome is annotated
# Update CARD database
rgi load --card_json ~/card_database/card.json
# Try --include_loose for more hits
rgi main --include_loose ...
# Use all three tools
run_dbcan --tools all ...
# Check protein quality
grep ">" proteins.faa | wc -l
| Feature | Typical Range |
|---|---|
| BGCs | 5-30 |
| AMR genes | 0-50 |
| CAZymes | 50-500 |
| Prophages | 0-10 |
| CRISPR arrays | 0-5 |
| Integrons | 0-5 |
What it does:
● Visualizes antiSMASH BGC distributions across genomes ● Creates 3 heatmap variants (basic, custom colors, annotated) ● Includes toy data built-in ● Automatic clustering by BGC similarity
Outputs:
● bgc_heatmap_basic.pdf - Simple clustered heatmap ● bgc_heatmap_custom.pdf - Custom gradient (white → gold → red → purple) ● bgc_heatmap_annotated.pdf - With annotations for BGC richness ● bgc_summary.csv - Summary statistics
Toy data included: 15 genomes × 9 BGC types
What it does:
● Creates bubble plots showing CAZyme family distributions ● Bubble size = CAZyme count ● Color = Genome completion percentage ● Includes heatmap and bar plot variants ● Analyzes degradation capabilities
Outputs:
● cazyme_bubble_plot.pdf - Main bubble visualization ● cazyme_bubble_plot_alt.pdf - Alternative color scheme ● cazyme_total_barplot.pdf - Total counts by taxonomic order ● cazyme_heatmap.pdf - Clustered heatmap ● cazyme_summary.csv - Summary with degradation analysis
Toy data included: 12 taxonomic orders × 6 CAZyme families
All R-codes available in *Day 6 →
| Tool | Single Genome | 10 Genomes |
|---|---|---|
| antiSMASH | 30-60 min | 8-10 hrs |
| RGI | 5-10 min | 1-2 hrs |
| ABRicate | 1-2 min | 15-20 min |
| dbCAN | 10-20 min | 2-3 hrs |
| VirSorter2 | 20-40 min | 5-7 hrs |
| InterProScan | 1-2 hrs | 15-20 hrs |
Congratulations! You’ve discovered the specialized capabilities of your genomes!
Next steps:
🔗 Day 6 →
Last updated: February 2026